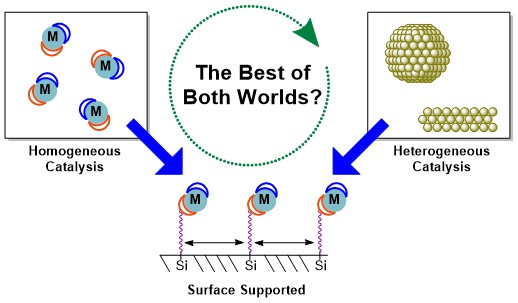
Introduction
Catalysts participate in chemical reactions to provide a lower energy pathway and are regenerated at the end of the process.1 They achieve this by lowering the activation energy, which is usually observed as a decrease in the required temperature or pressure. In some cases, reactions that are not possible without a catalyst become possible when one is present. An example is the hydrogenation of a ketone to form an alcohol (Scheme 1a).2 Ketones do not react directly with H2, but a palladium catalyst (Pd on carbon) mediates the reaction between dihydrogen and the ketone so it can occur at room temperature.

Scheme 1. Comparison of a) the catalytic reduction using H2 and a palladium catalyst and b) stoichiometric reduction of a ketone using NaBH4. The atom economy of the catalytic reaction is 100% and the stoichiometric reaction has an atom economy of 81%.
Catalysts can also remove the need for stoichiometric reagents. To reduce a ketone without a catalyst, a reducing agent (e.g. NaBH4) is necessary, which creates stoichiometric amounts of H3BO3 and NaOH byproducts (Scheme 1b). The catalyst, however, removes the need for these reagents and byproducts, which improves the efficiency of the reaction. This can be quantified by calculating the atom economy of the reaction, which is the percentage of atoms in the starting materials found in the products.3 In the hydrogenation of a ketone, this improves from 81% to 100% when moving from using stoichiometric reducing agents to a catalytic process. Usually, the stoichiometric reaction is even worse than this theoretically possible case as excess reducing agent is often used. Theoretically, since the catalyst is not changed during the reaction, it can be reused multiple times.
Due to these factors, catalysis is one of the 12 Principles of Green Chemistry and is the subject of much academic and industrial research.4 Catalysts are used in the production of over 90% of commercial chemicals, since along with decreasing waste, they also decrease the costs of reactions. With the global market for catalysis valued at 52.8 billion NZD in 2023, there is always interest in improving catalytic methodology by making existing catalysts more efficient, or increasing the scope of industrially viable catalytic reactions.5
Another major advantage of catalysts is that they can induce product selectivity in a reaction. By decreasing the relative energy of the formation of one product over another, the yield of the desired product can be increased. Hydroformylation (also known as the Oxo process) is a good example of this.6–9 In this reaction, a hydrogen atom and formyl group (-CHO) are added across an alkene to form an aldehyde product (Scheme 2). Depending on the direction of addition, either a linear or branched product are made. The linear products have a higher value, so catalysts are used in industrial processes to favour their formation. If selectivity of 100% can be achieved, the purification costs of separating the branched products from linear byproducts would be eliminated.
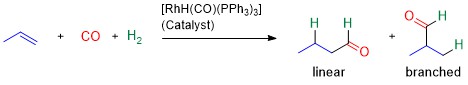
Scheme 2. Hydroformylation reaction using a rhodium catalyst to synthesise aldehydes from alkenes. Two products are possible: linear and branched.
Classes of catalyst
Catalysts fall under two broad categories, which depends on their phase relative to the reagents.10 Homogeneous catalysts are in the same phase as their reagents, usually solution, such as the hydroformylation catalyst [RhH(CO)(PPh3)3]. In contrast, heterogeneous catalysts are in a different phase to reagents. In such cases, the catalysts are almost exclusively solids, and the reagents are solutions, liquids or gases. Common examples include catalytic convertors in cars or palladium on carbon (Pd/C) for hydrogenation reactions. Nanoparticles are another example of heterogeneous catalysts, which are clusters of metal atoms (Scheme 3a).11
Heterogeneous catalysts are used extensively in industry to make many products from plastics to ammonia (for fertiliser).12,13 As heterogeneous catalysts are solids that do not dissolve in the reaction medium, they are easy and cheap to separate from the products and can be used in batch or flow processes. Due to the nature of their structures, they are often stable at high temperatures and pressures, can be reused multiple times and even regenerated if their activity starts to decrease. Heterogeneous catalysts do, however, have several disadvantages. The active sites of these catalysts (where the reactions occur) are often very difficult to locate and precisely characterise.14 Catalysts contain multiple possible active sites such as a particular face or edge of the solid, or even in a site created by a defect. Consequently, mechanistic information about how the reactions proceed is often lacking. Furthermore, there is typically poor synthetic control over tuning or adapting the active site to improve the activity or selectivity of the catalyst. Mechanisms are commonly drawn as schematics as seen in Scheme 3b.15
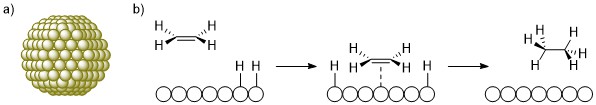
Scheme 3. Representative structure and mechanism for a heterogeneous catalyst; a) schematic of a nanoparticle, which is a cluster of metal atoms; b) schematic mechanism of hydrogenation of ethene with H2 on Pd heterogeneous catalyst.
In contrast, homogeneous catalysts (in the same phase as reagents) are usually easier to characterise and investigate.16 There are a wide variety of types of homogeneous catalyst, from organocatalysts to coordination complexes. In this discussion, we will focus exclusively on transition metal coordination complexes as catalysts. These consist of a transition metal ion surrounded by ligands (see Scheme 4).17 The catalytic mechanism for a coordination compound [RhH(CO)(PPh3)3] performing hydroformylation is shown in Scheme 4a.18 Here, precise steps can be proposed and the point determining the selectivity of the reaction identified, which can inform improvements to the catalyst structure to increase rate or selectivity for the desired products.
Ligands directly impact the reactivity of a coordination complex by altering the steric environment and electronic nature of the transition metal ion. Changing the ligands to be more electron donating or withdrawing can better stabilise the metal during key transition states, increasing the rate of the reaction or stability of the catalyst. Similarly, ligand size can be increased or decreased in size to select for the formation of a particular product. For example, chiral ligands (Scheme 4b) can promote the selective formation of a specific enantiomer product, which can be particularly useful in the synthesis of pharmaceuticals.19,20 Some ligands are “non-innocent” and actively take part in the reactions, often by being oxidised or reduced in place of the metal (Scheme 4c).21

Scheme 4. The well-defined structures and mechanisms for homogeneous catalysis; a) mechanism of hydroformylation to form a linear product using a rhodium catalyst – the direction of migratory insertion step labelled A determines whether the branched or linear product is formed; b) a Ni catalyst for asymmetric hydrogenation – chirality is induced in the product by the ligand; c) non-innocent ligand being reduced in place of the Fe(II).
The characterisation of homogeneous compounds is also easier, using techniques such as multinuclear NMR spectroscopy and X-ray crystallography to determine the nature of the metal-ligand bonds and how reagents interact with the metal. Nonetheless, it is still challenging to determine the exact catalytically active species as they are highly reactive and often short lived. Therefore, studies involving stoichiometric reactions, kinetic analysis (by studying the rates of reaction) or computational modelling are conducted before a catalytic cycle is proposed.
Homogeneous catalysts are rarely used in industrial processes.22 There are two main reasons for this: cost and practicality. The ligands used in homogeneous catalysis are often expensive and the nature of the catalyst more delicate relative to heterogeneous catalysts. This higher sensitivity is derived from the often highly reactive active catalytic species that is not stable for long periods or under harsh conditions such as high temperatures and pressures. This leads to decomposition of the catalyst so it cannot be reused and must be replaced. Since transition metal catalysts often contain rare metals such as platinum or rhodium, this cost is significant.
In industry, further costs are associated with the practicalities of using homogeneous catalysts in large scale processes. As homogeneous catalysts are in the same phase as the reaction products, purifying the products and regaining the catalyst requires costly separation processes such as distillation, crystallisation or chromatography. This is a particular challenge in the pharmaceutical industry where traces of toxic metals in the products are unacceptable. The process to remove a catalyst can also change its structure, making it more difficult or impossible to recycle and reuse, further increasing costs. Although improvements are being made in these areas, heterogeneous catalysts are still preferred.23,24
There are occasions when homogeneous catalysts are used in industry.16 To be economically viable, advantages such as the formation of a higher value product, decreased byproducts or stoichiometric reagents needs to outweigh the cost of removing the catalyst from the products. Examples of homogeneous catalysts in industry include on a small scale for specialised fine chemical products (e.g. pharmaceuticals) or on a large scale for the selective formation of linear products in hydroformylation. These are, however, the minority. Given the ~900 academic articles published every year on new homogeneous catalytic methodology, there is a significant disconnect between this and the very few examples that make it into industrial processes.
Attaching catalysts onto solid supports
One solution is to affix homogeneous catalysts onto solid supports. This process, often called “immobilisation” attaches transition metal coordination compounds onto the surfaces of solids such as silica (SiO2), titania (TiO2) or alumina (Al2O3) to convert them from homogeneous to heterogeneous catalysts.25 In an ideal case, the resulting “solid-supported catalysts” combine the advantages of homogeneous and heterogeneous catalysis to make highly selective catalysts that can be easily tuned for the desired reactivity, cheaply removed from the reaction products and recycled multiple times. This concept has been explored since the 1970s but recent developments continue to garner interest in this topic including new support materials, and improved synthesis and characterisation methods.
Inspiration for attaching homogeneous catalysts to solid supports can be found in heterogeneous catalysts, as these are usually attached to or dispersed on inert supports such as inorganic solids Al2O3 and MgCl2.26 Multiple techniques are known depending on the catalyst structure. In general, these techniques are used to increase the surface area of the heterogeneous catalyst. For example, thermal deposition disperses a fine layer of metal catalyst over an inert support. Since usually only the surface layers of the catalyst react, this method increases the amount of reactive metal, relative to using the bulk metal. In the case of metal nanoparticles, affixing them to solid supports also increases thermal stability relative to using unsupported nanoparticles.27 None of these methods, however, overcome the difficulties of using heterogeneous catalysts as they all result in a mixture of potential active sites from which it is challenging to identify which are responsible for catalysis, or tune them for better reactivity.
To make more controllable systems that contain uniform metal sites, catalysts attached onto the solid supports need to maintain discrete coordination compounds with identical structures. If the system is simplified completely, we reach the extreme of single atom catalysts.28,29 Here, discrete atoms are dispersed on a support (Scheme 5). Examples include platinum atoms on iron oxide supports (Pt1/FeOx) for CO oxidation.30

Scheme 5. Single atom catalysts attached to solid support. High temperatures can cause decomposition by migration of the atoms into clusters or nanoparticles.
Since these atoms are attached only to the solid support, they are inherently highly reactive (due to their low coordination number). This also makes them unstable to higher temperatures and harsh conditions. Common decomposition routes include the migration of these single atoms to form nanoparticles, once again forming a heterogeneous catalyst with a variety of metal environments.
Single atom catalysts also lack methods for modifying their reactivity, since only the support can be changed. Therefore, there is very little control over their reactivity or selectivity in a particular reaction. The key difference between single atom and homogeneous catalysts is the ligand.
Surface organometallic chemistry (SOMC) attaches well-defined homogeneous coordination complexes onto solid supports.31 Its aim is to fully mimic the control exhibited in homogeneous catalysis whilst attached to a solid support.
Solid supports
When considering attaching a catalyst onto a support, it is important to choose the best support for the application. The support itself greatly impacts the mode of attachment, concentration of catalyst and catalyst structure. In general, supports with a high surface area are used, as they increase the amount of catalyst per gram. For example, “mesoporous” silica (Fig. 1) contains ordered pores running through its structure and can achieve surface areas of up to 1000 m2/g.32 Catalysts attached to these high surface area supports will have a much higher amount of metal per gram compared to when attached to amorphous silica with a surface area of 50 – 380 m2/g. This has practical advantages in boosting spectroscopic signals during characterisation and lowering the required mass of catalyst that needs to be added to a reaction.

Fig. 1. a) Scanning transmission electron microscopy (STEM) image of SBA-15, a mesoporous silica with hexagonal pores that run through the material; b) schematic showing structure of the pores.
It is also important to consider how evenly the metal coordination complexes are dispersed on the solid support (Fig. 2). If the dispersion is poor, areas of high concentration will occur, where many metal complexes will be clustered together. This increases the probability of reactions between metal coordination complexes, which can lead to multiple different species attached to the surface. Furthermore, reactions between transition metal catalysts often results in deactivation, as can be observed in mechanistic investigations of homogeneous catalysis.33 Solid-supported catalysts that achieve even distribution and avoid reactions between metal catalysts can exhibit even better reactivity and longevity than analogous homogeneous catalysts as decomposition pathways are avoided.
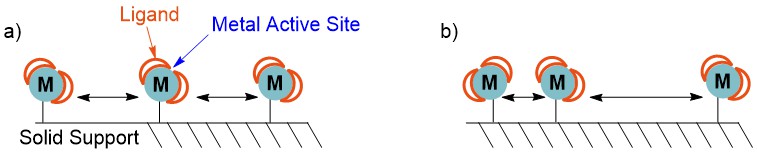
Fig. 2. Distribution of transition metal coordination compounds on the surface of solid supports; a) even, uniform distribution where metal centres are too far apart to react with each other; b) uneven distribution where some coordination compounds can react together.
The method of attachment to the support is also important. Coordination compounds can be attached to solid supports by physisorption and chemisorption.34 In physisorption, Van der Waals forces bind the catalyst to the support. These forces are weak and easily broken, especially at higher temperatures. Coordination compounds attached using physisorption are more likely to migrate on the support to form clusters or nanoparticles, losing their well-defined nature. When catalysts are lost into solution (leaching), the catalyst will become less active over time and the products are contaminated.
Chemisorption occurs when chemical bonds (covalent or ionic) are made between the catalyst and support (Fig. 3). Covalent bonds are strong and not as easily broken, so this usually results in the catalyst being firmly anchored to the support. This does, of course, depend on the strength of bond formed. For example, early transition metals form stronger covalent bonds with oxygen than late transition metals. Therefore, when transition metals are directly attached to silica (SiO2) via Si–O–M bonds (Fig. 3b), early transition metals (e.g. M = Ti) are attached more firmly than late transition metals (e.g. M = Ir). Chemisorption through ionic bonds is less common and requires a charged support (Fig. 3a).35 If the catalyst changes its charge and becomes neutral during the reaction, this would result in the loss of the ionic bond and the catalyst being weakly physisorbed to the support.
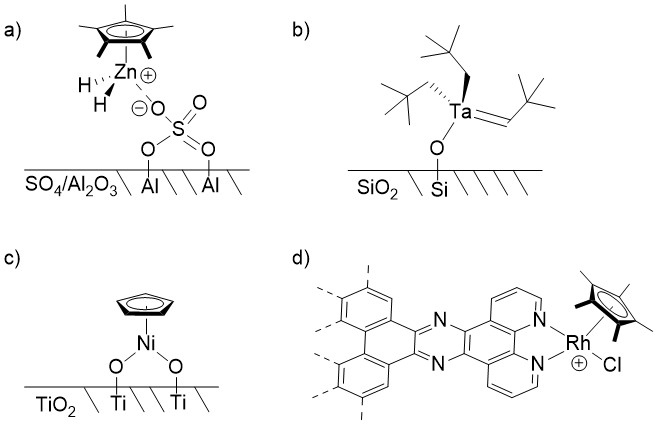
Fig. 3. Surface immobilised transition metal coordination compounds affixed a) by an ionic bond, b) by a covalent bond, c) to a redox active support, d) to a graphite electrode.
Strong chemisorption does not stop the formation of aggregates or nanoparticles, as this can result from poorly dispersed metal coordination compounds reacting together.
Finally, the reactivity of the support should be considered. Once again, two main classes exist: inert and reactive supports. The latter class contains supports that can take part in the catalytic reactions. For example, redox active supports can be oxidised or reduced during catalysis, in much the same way as non-innocent ligands (Fig. 3c).36 Or alternatively, conductive supports such as graphite or gold electrodes can directly pass electrons to the catalyst to perform electrocatalysis (Fig. 3d).37
Inert supports are the most widely studied, with silica (SiO2) being the most common. Since these supports do not take part in the reactions, this simplifies the system and the support itself does not need to be included in the catalytic mechanism. Although superficially similar, alumina (Al2O3) is a more challenging support to work with as the atoms are more mobile and can migrate to alter the connectivity between catalyst and support.38
Polymers are another type of inert support, where ligands can be placed periodically along the length of the polymer (Fig. 4a).39 It is worth considering that polymers have more flexible structures and so even if the coordination complexes are dispersed along the chain of the polymer, it could fold to bring them closer together than initially designed (Fig. 4b).
Metal organic frameworks (MOFs) are another increasingly common support for catalysts. These high surface area, porous networks are made of metal clusters connected by organic ligands. If these ligands have extra donor sites, they can also be used to bind to transition metals to make coordination complexes and perform catalysis (Fig. 4c).40,41

Fig. 4. a) Polymer supported Ir catalyst;39 b) catalysts attached to polymers may be closer together than designed if the polymer can fold or crosslink; c) organic linker for a MOF that can act as a ligand for various metals.
Silica is by far the most investigated support due to its generally inert nature and various methodologies to synthesise different structures. As previously described, a high surface area is desirable in solid supports for catalysis. Mesoporous silica has a high surface area due to it ordered pores (2-50 nm in diameter) that are made by using a template during synthesis.32 The size of the pores and surface area can be altered during synthesis by changing the size of the templating agent, pH and temperature. A larger pore size would decrease overall surface area but would increase diffusion rates into and out of the pores. Large pores may be desirable when installing a catalyst with large ligands or attempting to react with large reagents. The two most common types of mesoporous silica are SBA-15 (Fig. 1) and MCM-41, which both have hexagonal arrays of pores.
Multiple methods are available to attach coordination compounds to the silica, which vary in how easy they are to achieve synthetically and the structure of the resultant catalyst. Three methods are discussed in turn below, which demonstrate the considerations needed when attempting to attach a homogeneous coordination compound to a solid support for catalysis.
Method 1: Direct attachment to silica
The surface of silica is covered with silanol (Si–OH) groups, which are in various environments: vicinal, geminal or isolated (Scheme 6 inset). These silanol groups can be used to attach to transition metal complexes by forming Si–O–M covalent bonds directly from the silica to the desired transition metal (M).31 If this were attempted using unmodified silica that contains all types of silanol groups (vicinal, geminal and isolated), the metal complexes formed would be in a variety of different environments and poorly dispersed.
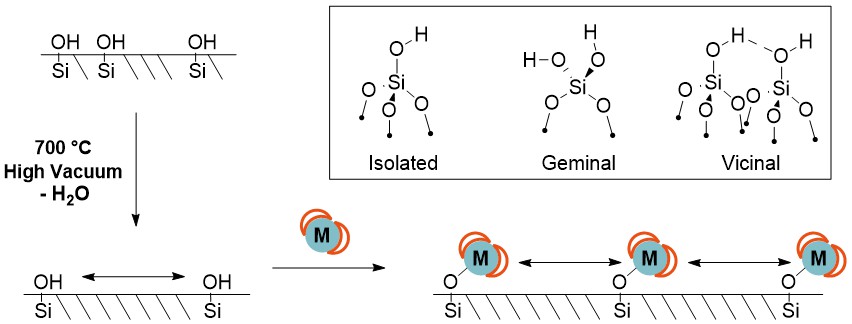
Scheme 6. Schematic of attaching organometallic compounds direct to silica. The silica surface is functionalised with many different types of silanol (inset). Removal of water using high temperature creates dehydroxylated silica with only isolated silanol groups. Metal coordination compounds can be attached via Si–O–M bonds.
Consequently, silica supports are often treated before metal coordination complexes are attached or “grafted” onto the surface. Dehydroxylating the silica using high temperatures (200 to 700 °C) under high vacuum (10-6 mbar) decreases the concentration of silanol groups on the silica surface. At higher temperatures, more water is removed and the concentration of silanol groups decreases. For example, silica dehydrated at 700 °C (e.g. SBA-15700) contains only isolated silanol groups.31 Grafting metal coordination compounds to this silica results in uniformly dispersed metal species that are spread far enough apart that they cannot interact with each other.
When grafting a metal coordination complex directly onto silica, the coordination environment of the metal complex will change. Usually, this reaction proceeds via protonolysis, where the proton of the silanol group cleaves a metal-ligand bond (Scheme 7). This results in the metal coordination complex losing a ligand, which is then replaced by the Si–O–M bond. This has two consequences: a ligand is lost from a catalyst, and it is replaced by a silanol group. If the lost ligand was important for catalysis, the activity or selectivity will be dramatically affected. Coordination complexes can be designed so important ligands are not lost during this step but sometimes this is not possible, decreasing the scope of catalysts compatible with immobilisation by this method.
It can be very useful for characterisation if the ligand lost during the grafting reaction is quantifiable by GCMS or NMR spectroscopy, since this can determine the amount of metal complex attached to the silica. Alternatively, ICP-MS can determine the weight percentage (wt%) of metal, which is used to calculate the catalyst loading during subsequent reactions.
Post grafting functionalisation of the catalyst is also possible, and is indeed common, to remove further unwanted ligands. For example, hydrogenation of the mixed metal tantalum/iridium complex shown in Scheme 7 replaces an alkyl ligand with a hydride.42 When this hydrogenation occurs in solution, a dimer is formed, but when grafted onto silica it always remains as a monomer since the metal complexes are distributed far apart and cannot react together. This silica-supported version has superior catalytic activity in C-H activation due to its smaller substituents.
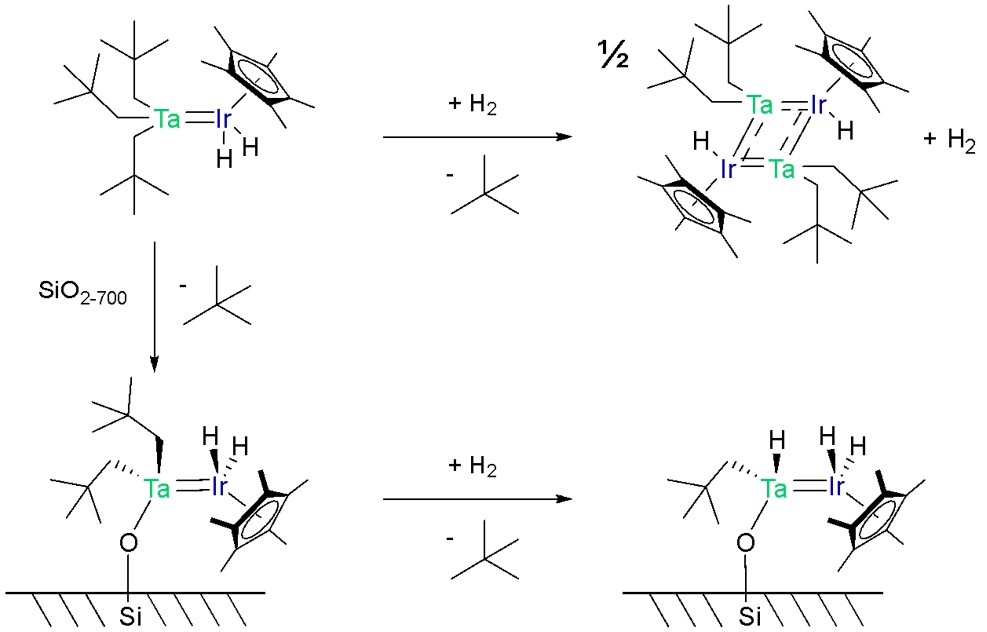
Scheme 7. Top: Hydrogenating a Ta/Ir coordination compound results in loss of an alkyl ligand and dimerisation. Left: Grafting it to silica dehydroxylated at 700 °C results in loss of one alkyl ligand. Bottom: one more alkyl ligand can be lost by hydrogenating, but no dimerisation occurs as the coordination compounds are spread too far apart to react.
Several other exotic compounds have been synthesised grafted onto silica, though it is worth noting here that sometimes in the absence of other species to react with, the metal may react with the support. An example is the tantalum alkyl complex grafted onto silica shown in Fig. 3b, which in the presence of hydrogen becomes a highly reactive tantalum hydride.43 This breaks open an Si–O–Si bond to form an extra Si–O–Ta bond.
Overall, this method is useful to create highly reactive metal complexes with small substituents and has been reported extensively by those working in the field of surface organometallic chemistry. There are, however, some disadvantages. In particular, the formation of an Si–O–M bond, which may not be optimal for catalytic behaviour. This method also requires specialised equipment to prepare the silica, such as access to a high vacuum setup, furnace and quartz glassware to dehydrate silica at 700 °C. Finally, it was already mentioned that some metals are more suitable for attaching directly to silica than others. Early transition metals will bind strongly with the silanol ligands, whereas late transition metals are poorly anchored and will be more easily leached from the surface, or migrate to form nanoparticles.
Method 2: Attaching silanols with ligands
A second method of attaching metal coordination compounds onto silica does not require this specialised equipment but does require the synthesis of a specialised ligand. In this method, a ligand is attached to a silanol group, which is then grafted onto the silica support via a condensation reaction that forms new Si–O–Si bonds (Scheme 8).44 No prior treatment of the silica is necessary and the grafting procedure itself simply involves stirring the desired silanol with the silica. This process affixes the silanol onto the surface of the silica and the ligand can then coordinate to the desired metal. As well as the ligand and metal, the organic linker between ligand and silanol can also be altered (e.g. length or flexibility), which may impact how the catalyst reacts.45
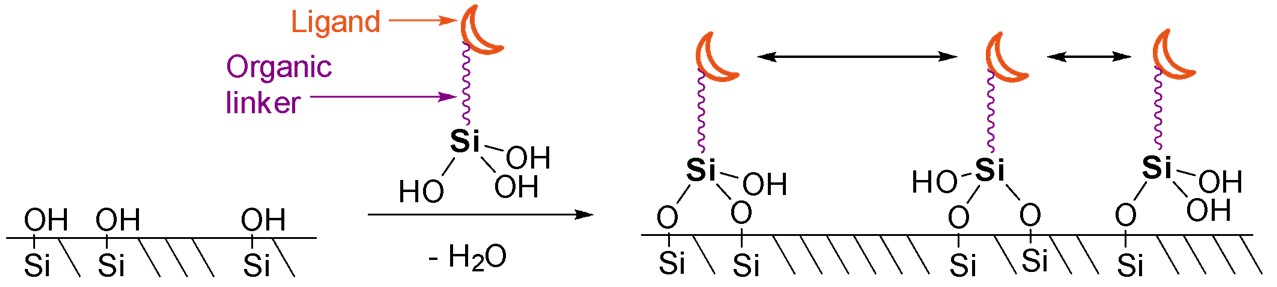
Scheme 8. Grafting a silanol affixed to a ligand onto silica via a condensation reaction. Uneven distribution on the surface occurs.
This method has been widely reported for a variety of metal coordination complexes and their resultant catalysis. For example, a silanol with attached BINAP ligand was synthesised, grafted to silica and then coordinated to rhodium and ruthenium complexes for asymmetric hydrogenation reactions.46
There are a few disadvantages to this method. First, the synthesis of the silanol with ligand attached can be non-trivial. In the above BINAP example, the silanol synthesis required 3 steps and the authors admit that it was not completely clean of impurities before they grafted it onto silica.
Another disadvantage is the strength of the connection between silica and grafted silanol. Although theoretically, three new Si–O–Si bonds could be formed, in reality only one or two are made.47 This results in relatively poor anchoring of the silanol, its attached ligand and metal catalyst onto the silica and makes leaching of the metal into solution during the reaction more likely. Once this occurs, the products will be contaminated and the catalyst will become less effective over time.
There is also a problem with distribution of catalyst across the silica. With a mesoporous silica that contains pores, grafted silanes cluster around the entrance to pores and do not evenly distribute themselves across the surface and into the pores.48,47 This results in areas of high concentration of catalyst, which can then react with each other, forming a variety of metal environments or catalyst deactivation.
Finally, the selectivity of metal binding should be considered. Once the silanol and ligand have been grafted onto the silica, any metal to be coordinated can now either coordinate to the ligand (as is desired), or it could bind directly with any remaining silanol groups on the silica surface (Scheme 9). A step called “passivation” is necessary, which adds a protecting group such as methyl, ethyl or trimethylsilyl onto any remaining surface silanol groups.49 The ligands themselves must be robust to these passivation conditions or they may not be able to bind to the metal afterwards. Alarmingly, many literature examples often omit this passivation step, resulting in competition for coordination of the metal and significant non-uniformity in the metal coordination environments.
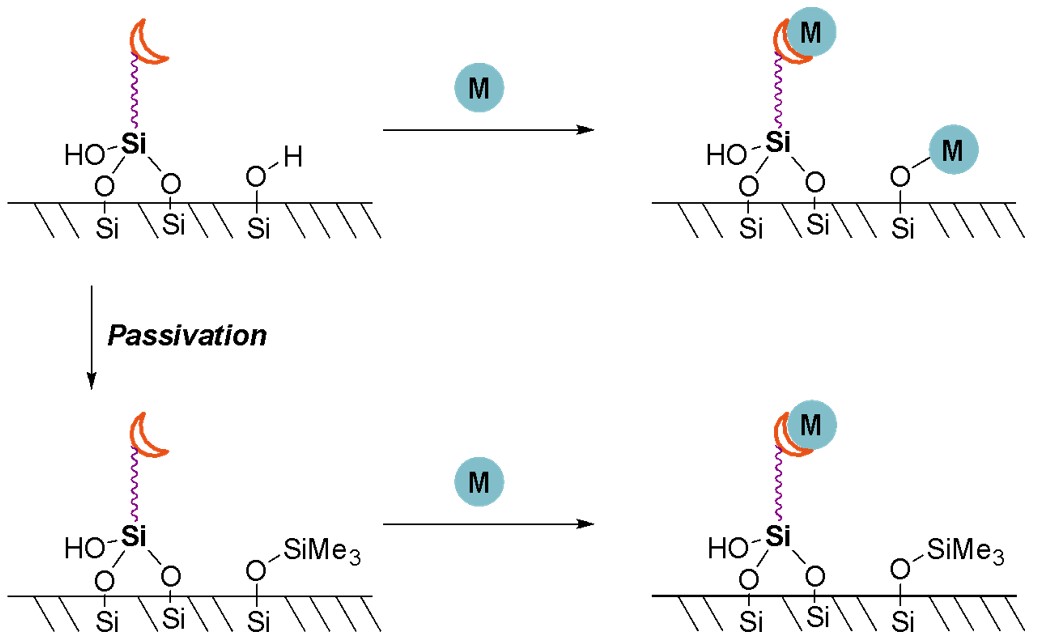
Scheme 9. Coordination of a metal to a silica affixed ligand. Top: with no passivation, the metal may coordinate to the ligand and also to the surface silanol groups. Bottom: If silanol groups are passivated (protected), selective coordination to the ligands is possible.
Overall, this method has several advantages over the first including that it is easier to perform and that the Si–O–M ligand can be replaced with a more desirable or catalytically appropriate ligand. There are also several disadvantages such as the poor anchoring of the catalyst to the silica and the uneven distribution on the silica surface. Despite this being the most popular choice in the literature, examples are often poorly considered and do not take into account the possible products such as the metal coordinating to the silica surface instead of the installed ligand.
Method 3: Hybrid silicas
Hybrid silicas contain a mixture of silanol groups and functional groups on their surfaces. (Scheme 10).49 These functional groups are installed by adding functionalised silanols during the silica synthesis. The resultant hybrid materials can be made in a variety of structures including the mesoporous high surface area silicas previously described, e.g. SBA-15. The functional groups are firmly anchored into the silica and less likely to be lost than those attached via the silanol grafting method described in Method 2.

Scheme 10. Synthesis of mesoporous hybrid silica with iodopropyl groups on the surface. The ratio of functonalised to unfunctionalised silanol dictates how far apart the functionality is dispersed on the surface of the silica product. The templating agent is responsible for the porous structure of mesoporous silica.
During the synthesis, it is key that the functionalised silanols are fully distributed amongst the unfunctionalised silanols so they are uniformly dispersed throughout the resultant silica. Hybrid silicas have been synthesised with a limited number of functional groups since some functionalised silanes do not distribute well during the synthesis. For example, silicas featuring iodopropyl groups have been synthesised with a ratio of 1 silanol to 19 or 30 iodopropylsilanols but amino functionalised hybrid silicas are more challenging to synthesise.50,51
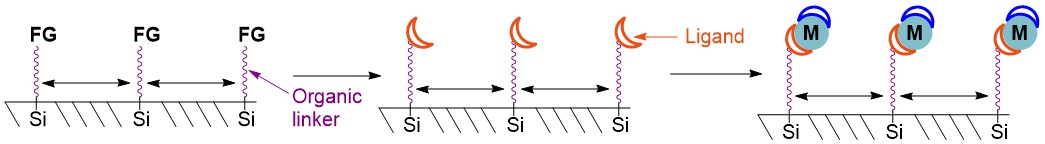
Scheme 11. Hybrid silica with functional groups (FG) distributed evenly and attached firmly to the silica surface. Conversion of the functional groups to ligands and attachment of metal coordination compounds.
Simple reactions are then used to convert the original functionality into the desired ligand, followed by metal coordination (Scheme 11). The higher the proportion of unfunctionalised silanol in the synthesis, the further apart the resultant catalyst will be on the support surface.
Although silica is robust to many reaction conditions and solvents, care should be taken when designing synthetic routes. Since many purification methods are not possible on the silica (e.g. crystallisation, distillation), the reactions should be high yielding, fast and be easily separated from their side products. Examples include the synthesis of N-heterocyclic carbene ligands for use in alkene metathesis catalysis, but few ligands have been synthesised by this method, so the scope is currently limited.45,52–54 Finally, passivation of the remaining surface silanols is necessary before adding the final metal coordination compound to synthesise the desired catalyst.
Although synthetically more challenging than the silanol grafting method, the resultant silica-supported catalysts have more controllable structures with better dispersion of catalysts across the silica surface. This more sophisticated version results in catalysts that cannot interact with or deactivate one another and are better anchored to the silica surface to decrease the likelihood of leaching metal and contaminating the products. The resultant catalysts closely mimic homogeneous catalysts with little change in the catalyst structure, resulting in comparable or improved catalytic activity.
Conclusions
In conclusion, affixing homogeneous catalysts onto solid supports can convert them into heterogeneous catalysts. Using this method, activity and longevity of the catalyst can be enhanced compared to homogeneous catalysts. For example, when coordination complexes are dispersed evenly onto solid supports, this stops reactions between catalyst molecules, thereby removing some catalyst deactivation pathways such as nanoparticle formation. Ligand size can also be decreased, leading to higher reactivity with substrates and in some cases very unusual chemical motifs. Catalysts can feature cooperation between the coordination complex and the support, including for electrochemistry, enhancing the reactivity over homogeneous catalysis alone.
Other methods, however, lead to a poor distribution of catalysts on the support, which can then react together and make multiple species on the surface. This removes the advantages afforded by homogeneous catalysis and makes them more akin to heterogeneous catalysts which are difficult to study and identify the reactive species.
The process of performing surface organometallic chemistry should, therefore, be approached with caution and careful considerations of which methodology to use rather than a silver bullet that will make any catalyst more applicable in industry.55 Indeed, one of the major issues with using homogeneous catalysts in industrial processes is the high cost of ligands. In surface organometallic chemistry, the solid support can be equally expensive to make, so it is important that the advantages afforded by immobilising the catalysts outweigh the costs. For example, applicability in flow processes, high recyclability and stability such that the catalyst can be recycled and reused multiple times.
Further uses for solid-supported catalysts are their applicability in mechanistic investigations such as kinetic analysis so their catalytic mechanisms can be determined. This can inform improvements to their structure by altering ligands in a way that is very challenging for heterogeneous catalysts.
Overall, with careful consideration, surface organometallic chemistry can have many uses and continues to develop to be a useful tool for academic and industrial research and applications alike.
References
- Roduner, E. Chemical Society Reviews, 2014, 43, 8226–8239.
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, Oxford University Press, Oxford, UK, 2012.
- https://chem.libretexts.org/Bookshelves/Environmental_Chemistry/Green_Chemistry_and_the_Ten_Commandments_of_Sustainability_(Manahan)/05%3A_Chemical_Reactions-_Making_Materials_Safely_and_Sustainable/5.04%3A_New_Page, (accessed 06/08/2025).
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice, Oxford University Press: New York, 1998.
- Catalyst Market Size & Trends Analysis Report, 2023, https://www.grandviewresearch.com/industry-analysis/catalyst-market, (accessed 05/08/2025).
- Adkins, H.; Krsek, G. Journal of the American Chemical Society, 1949, 71, 3051–3055.
- Evans, D.; Osborn, J.A.; Wilkinson, G. Journal of the Chemical Society A: Inorganic, Physical, Theoretical, 1968, 3133–3142.
- Franke, R.; Selent, D.; Börner, A. Chemical Reviews, 2012, 112, 5675–5732.
- Zhang, B.; Peña Fuentes, D.; Börner, A. Chem Texts, 2021, 8, 2.
- Types of Catalysis, https://www.chemguide.co.uk/physical/catalysis/introduction.html, (accessed 6 August 2025).
- Astruc, D. Chemical Reviews, 2020, 120, 461–463.
- Li, W.; Wang, J.; Yang, Y. ChemCatChem, 2024, 16, e202300812.
- The Haber Process, https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Equilibria/Le_Chateliers_Principle/The_Haber_Process, (accessed 06/08/2025).
- Sheldon, R.A.; Arends, I.W.C.E.; Hanefeld, U. In Green Chemistry and Catalysis, John Wiley & Sons, Ltd, 2007, 91–131.
- https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Inorganic_Chemistry_(LibreTexts)/14%3A_Organometallic_Reactions_and_Catalysis/14.04%3A_Heterogeneous_Catalysts, (accessed 05/08/2025).
- Crabtree, R.H. In The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Ltd, 2014, 224–258.
- Coordination Chemistry, https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Supplemental_Modules_and_Websites_(Inorganic_Chemistry)/Coordination_Chemistry, (accessed 06/08/2025).
- Elschenbroich, C. Organometallics, Wiley, 3rd ed, 2006.
- Li, B.; Wang, Z.; Luo, Y.; Wei, H.; Chen, J.; Liu, D.; Zhang, W. Nature Commun, 2024, 15, 5482.
- Zhang, L.; Meggers, E. Chemistry Society Reviews, 2025, 54, 1986–2005.
- Lyaskovskyy, V.; De Bruin, B. ACS Catalysis, 2012, 2, 270–279.
- Joshi, S.S.; Ranade, V.V. Industrial Catalytic Processes for Fine and Specialty Chemicals, Elsevier, 2016.
- Vougioukalakis, G.C. Chemistry – A European Journal, 2012, 18, 8868–8880.
- PGM recent publications, https://matthey.com/products-and-markets/pgms-and-circularity/pgm-markets/pgm-recent-publications, (accessed 6 August 2025).
- Benaglia, M.; Puglisi, A. Catalyst Immobilization, John Wiley & Sons, Ltd, 1st ed. 2020.
- Munnik, P.; De Jongh, P.E.; De Jong, K.P. Chemical Reviews., 2015, 115, 6687–6718.
- Kim, S.; Varga, G.; Seo, M.; Sápi, A.; Rácz, V.; Gómez-Pérez, J.F.; Sebők, D.; Lee, J.; Kukovecz, Á.; Kónya, Z. ACS Applied Nano Materials, 2021, 4, 4070–4076.
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Accounts of Chemical Research, 2013, 46, 1740–1748.
- Mitchell, S.; Pérez-Ramírez, J. Nature Communications, 2020, 11, 4302.
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Nature Chemistry, 2011, 3, 634–641.
- Copéret, C.C.; Comas-Vives, A.; Conley, M.P.; Estes, D.P.; Fedorov, A.; Mougel, V.; Nagae, H.; Núñ Ez-Zarur, F.N.; Zhizhko, P.A. Chemical Reviews, 2016, 116, 323–421.
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka B.F.; Stucky, G.D. Science, 1998, 279, 548–552.
- Nascimento, D.L.; Foscato, M.; Occhipinti, G.; Jensen, V.R.; Fogg, D.E. Journal of the American Chemical Society, 2021, 143, 11072–11079.
- Piermatti, O.; Abu-Reziq R.; Vaccaro, L. in Catalyst Immobilization, John Wiley & Sons, Ltd, 2020, 1–22.
- Witzke, R.J.; Chapovetsky, A.; Conley, M.P.; Kaphan, D.M.; and Delferro, M. ACS Catalysis, 2020, 10, 11822–11840.
- Fan, L.; Long, J.; Gu, Q.; Huang, H.; Lin, H.; Wang, X. Journal of Catalysis, 2014, 320, 147–159.
- Jackson, M.N.; Oh, S.; Kaminsky, C.J.; Chu, S.B.; Zhang, G.; Miller, J.T.; Surendranath, Y. Journal of the American Chemical Society, 2018, 140, 1004–1010.
- Conley, M.P.; Gao, J.; Huynh, W.; Rodriguez, J.; Samudrala, K.K. in Comprehensive Organometallic Chemistry IV, Elsevier, Oxford, 2022, 583–608.
- Broicher, C.; Foit, S.R.; Rose, M.; Hausoul, P.J.C.; Palkovits, R.;, ACS Catalysis, 2017, 7, 8413–8419.
- Xia, Q.; Li, Z.; Tan, C.; Liu, Y.; Gong, W.; Cui, Y. Journal of the American Chemical Society, 2017, 139, 8259–8266.
- Pascanu, V.; González Miera, G.; Inge, A.K.; Martín-Matute, B. Journal of the American Chemical Society, 2019, 141, 7223–7234.
- Lassalle, S.; Jabbour, R.; Schiltz, P.; Berruyer, P.; Todorova, T.K.; Veyre, L.; Gajan, D.; Lesage, A.; Thieuleux, C.; Camp, C. Journal of the American Chemical Society, 2019, 141, 19321–19335.
- Avenier, P.; Taoufik, M.; Lesage, A.; Solans-Monfort, X.; Baudouin, A.; de Mallmann, A.; Veyre, L.; Basset, J.-M.; Eisenstein, O.; Emsley, L.; Quadrelli, E.A. Science, 2007, 317, 1056–1060.
- Díaz, U.; Brunel, D.; Corma, A. Chemical Society Reviews, 2013, 42, 4083–4097.
- Renom-Carrasco, M.; Mania, P.; Sayah, R.; Veyre, L.; Occhipinti, G.; Gajan, D.; Lesage, A.; Jensen, V.R.; Thieuleux, C. Dalton Transactions, 2019, 48, 2886–2890.
- McDonald, A.R.; Müller, C.; Vogt, D.; van Klink, G.P.M.; van Koten, G. Green Chemistry, 2008, 10, 424–432.
- Lelli, M.; Gajan, D.; Lesage, A.; Caporini, M.A.; Vitzthum, V.; Miéville, P.; Héroguel, F.; Rascón, F.; Roussey, A.; Thieuleux, C.; Boualleg, M.; Veyre, L.; Bodenhausen, G.; Copéret, C.; Emsley, L. Journal of the American Chemical Society, 2011, 133, 2104–2107.
- Hicks, J.C.; Dabestani, R.; Buchanan, A.C.; Jones, C.W. Inorganica Chimica Acta, 2008, 361, 3024–3032.
- Conley, M.P.; Copéret, C.; Thieuleux, C. ACS Catalysis, 2014, 4, 1458–1469.
- Alauzun, J.; Mehdi, A.; Reyé, C.; Corriu, R. New Journal of Chemistry, 2007, 31, 911–915.
- Li, Y.; Xiong, W.; Wang, C.; Song, B.; Zhang, G. RSC Advances, 2016, 6, 53991–54000.
- Karamé, I.; Boualleg, M.; Camus, J.-M.M.; Maishal, T.K.K.; Alauzun, J.; Basset, J.-M.M.; Copéret, C.; Corriu, R.J.P.J.P.; Jeanneau, E.; Mehdi, A.; Reyé, C.; Veyre, L.; Thieuleux, C. Chemistry - A European Journal, 2009, 15, 11820–11823.
- Baffert, M.; Maishal, T.K.; Mathey, L.; Copéret, C.; Thieuleux, C. ChemSusChem, 2011, 4, 1762–1765.
- Conley, M.P.; Drost, R.M.; Baffert, M.; Gajan, D.; Elsevier, C.; Franks, W.T.; Oschkinat, H.; Veyre, L.; Zagdoun, A.; Rossini, A.; Lelli, M.; Lesage, A.; Casano, G.; Ouari, O.; Tordo, P.; Emsley, L.; Copéret, C.; Thieuleux, C. Chemistry – A European Journal, 2013, 19, 12234–12238.
- Hübner, S.; de Vries, J.G.; Farina, V. Advanced Synthesis & Catalysis, 2016, 358, 3–25.