


Introduction
In November 2022, new drinking water standards came into effect in New Zealand, under Section 47 of the Water Services Act 2021.1 These standards are regulated by the government agency, Taumata Arowai, who are tasked with ensuring all communities have access to safe drinking water. The standards set the maximum acceptable values (MAV) for microbiological, inorganic, organic and radiological determinands in all drinking water which is provided by suppliers in New Zealand. The 2022 update of the standards included a MAV for a previously unregulated compound, N-nitrosodimethylamine, commonly abbreviated to NDMA.
By introducing a MAV for NDMA, New Zealand is following the lead of the World Health Organisation, who have set a guideline value of 100 ng/L for NDMA in drinking water.2 With drinking water suppliers now required to screen for NDMA, analytical methods will be required in New Zealand testing labs that can achieve the low levels of detection directed by the standards. This article briefly summarises the health risks, prevalence and formation of NDMA before focusing on the analytical techniques available for quantifying NDMA in water at the regulated concentration - a limit of quantification (LOQ) of 0.0001 mg/L, or 100 parts per trillion (ppt).
Health risks
NDMA is part of a class of compounds known as N-nitrosamines where a nitroso group is bound to an amine (Fig. 1). Of all N-nitrosamines, NDMA is the most well-studied due to its wide occurrence in the environment.3 In recent times it has gained more attention for the discovery that it can, and does, form in pharmaceuticals products.4,5 However, its presence in treated water has been known for decades.6

The US Agency for Toxic Substances and Disease Registry recently published a comprehensive review of, and toxicological profile for, NDMA.7 This revealed that in addition to the already well-established studies of NDMA as a carcinogen (lung, liver, testicular, gastric, bladder, prostate, kidney cancers formed in animals), oral exposure to NDMA can cause various types of liver damage and even death at high enough doses (5 mg/kg per day). This is in agreement with the US EPA Integrated Risk Information System designating NDMA as class B2 – probable human carcinogen.8
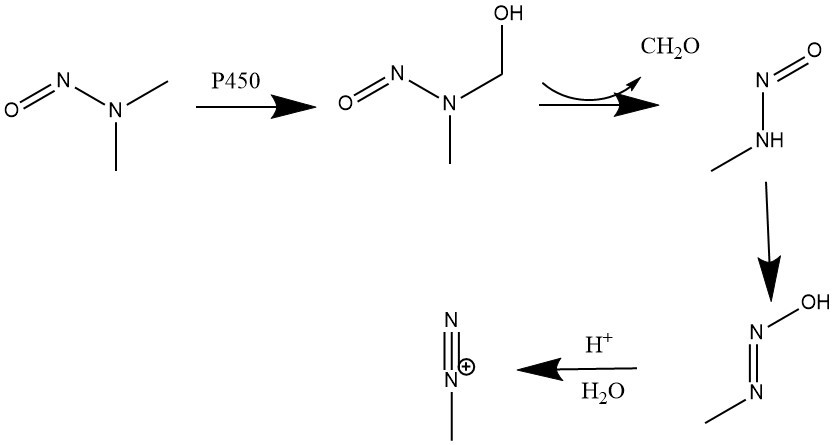
The mechanism by which NDMA causes cancer is through the cytochrome P450 enzyme. This interaction leads to α-hydroxylation of NDMA, forming an unstable dealkylated primary nitrosamine that degrades to diazonium (Fig. 2).9 Diazonium facilitates DNA alkylation, which can lead to formation of cancers.
As an emerging concern in dietary pathways, the European Food Safety Authority (EFSA) has recently established a lower confidence limit for the 10% benchmark dose (BMDL10) of 0.035 mg/kg bodyweight (bw) per day10 and that, based on a Margin of Exposure (MOE) approach there was a health concern from dietary exposure to N-nitrosamines in foods. This adds some support to the position of Taumata Arowai to regulate levels in NZ drinking water, and future changes to food safety legislation may follow suit.
Prevalence and formation in drinking water
The health risks of NDMA alone do not justify the requirement for drinking water suppliers to screen for it. To justify regulation, and the resource required to carry out testing, there must be a reasonable concern that it will occur in our drinking water.
While NDMA has been found in other environmental contexts, its ability to persist in water makes this the area of most concern. NDMA can accumulate in the indoor air of industrial sites manufacturing products such as pesticides, tyres and rubbers as well as in tanneries and fish processing facilities.11 In outdoor air, however, NDMA tends to degrade rapidly via photolysis.12,13 This degradation pathway is less prevalent in exposed water due to the presence of dissolved matter acting to screen NDMA from direct sunlight,14 with most water treatment systems closed to sunlight.
Over the last 25 years, literature has been published finding NDMA in drinking water across the world on every populated continent,15–21 including one study which found up to 74 ng/L in Australian treated waters.22
NDMA can form via many pathways in drinking water from a multitude of amine precursors.23 All the common industrial disinfection methods have been shown to yield NDMA if precursors are available, except for ultraviolet irradiation, which degrades NDMA by photolysis.24
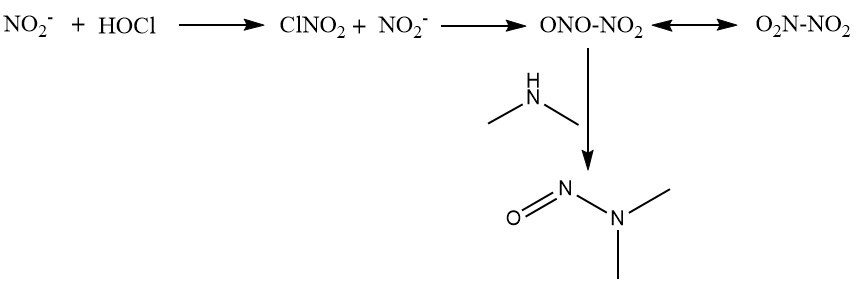
Chloramination is the most well studied source of NDMA formation as a disinfection by-product, due to it directly providing nitrogen compounds for the reaction.25 The amine precursor attacks the electrophilic dichloramine, which forms a hydrazine intermediate that undergoes oxidation to NDMA (Fig. 3).26
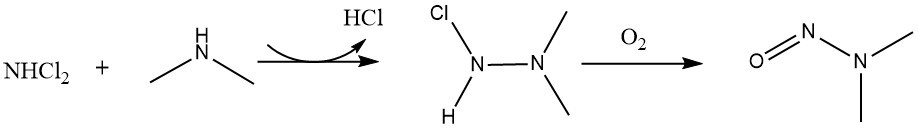
Chlorination by chlorine gas or sodium hypochlorite has also been shown to yield NDMA via formation of highly reactive nitrosylating intermediates (Fig. 4).27 This scheme has been modified from the original source to show the reaction proceeding via the correct isomer.28
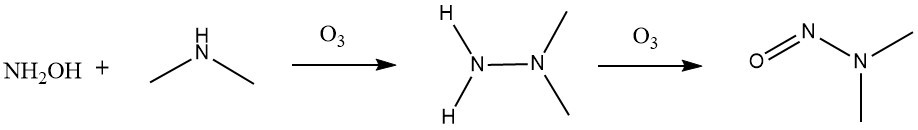
Ozonation of waters containing ammonia can lead to formation of hydroxylamine, which can subsequently react with amine precursors to form the hydrazine intermediate required for a final nitrosamine end product (Fig. 5).29
Analytical methodology
Due to the recent discovery that NDMA and other N-nitrosamines can form as contaminants during pharmaceutical manufacture, there has been a large increase in methods used to detect these compounds in drug matrices.30–34 While there are learnings that can be taken from these extraction and detection techniques, the scope of this review will focus on methodologies associated with water analysis.
The requirement for low detection limits of NDMA mean that methods must be specifically optimised to produce reliable quantitative results at the ppt scale.
Sampling
There is a requirement for care in how samples are taken for testing to minimise compound degradation and contamination. The US EPA gives excellent guidance on how to do this in Method 521 for N-nitrosamine analysis.35 The method describes the importance of taking field blanks, dechlorination, chilling the sample immediately and protecting it from light. The utmost care is taken to preserve the integrity of the sample, however, NDMA is not completely fragile. The chemistry of NDMA makes it less prone to degradation under mild aqueous conditions.36 This contributes to its persistence in natural or municipal waters, but also makes it stable as an analytical standard when dissolved in aqueous solutions, so long as it is protected from light. In fact, the EPA method found that NDMA in aqueous solution is stable for at least 45 days when stored at 4°C and protected from light.
Extraction
To achieve the highest possible sensitivity of the analytical method, an extraction or pre-concentration step is almost always included for NDMA analysis. There are advantages and disadvantages to each technique.
Liquid-liquid extraction
The use of liquid–liquid extraction for NDMA is limited because of its hydrophilic nature. It does not readily partition into organic solvent from an aqueous phase (log Poctanol–water -0.57).37 The high volumes of extraction solvents required and the subsequent concentration steps lead to variable recoveries and limits of detection (LODs) that are much higher than other available techniques.38 One study did, however, report the use of a micro-liquid–liquid extraction technique to overcome these limitations and still meet the sensitivity required by regulatory bodies.39 In addition, by utilising a large volume injection protocol for gas chromatography, Hu et al. achieved sub-ppt LODs with a continuous liquid–liquid extraction method.40 Overall, liquid–liquid extraction is largely irrelevant for extraction of NDMA from water samples, although it is vitally important for the detection of NDMA in the pharmaceutical industry.30
Solid-phase extraction (SPE)
For detection of NDMA in water at the required ppt levels, the most common sample preparation technique is solid-phase extraction (SPE).25 SPE is applied in NDMA analysis to increase the concentration of the analyte in solution. The water sample is passed through a tightly packed column of a sorbent which binds NDMA. The NDMA is then eluted in a smaller volume of solvent, that can be evaporated for further concentration. SPE traditionally follows five steps:41
- Conditioning the sorbent to activate the surface for interaction with the analyte. This step also helps remove any impurities from the sorbent manufacturing process, which is of particular importance when performing any kind of trace analysis.
- Equilibrating the sorbent to the same conditions as the sample. This is critical to having good reproducibility and recovery.
- Loading the sample onto the column of sorbent for selective retention of the analyte. When using SPE for concentration, it is important in this step to understand what capacity your SPE cartridge has for the matrix you are putting through, i.e., how much of the analyte can the sorbent retain before it starts passing straight through? Often, vacuum assisted flowrate is optimised for this step to provide maximum recovery.
- Washing the sorbent with a selected solvent to remove any undesirable retained compounds.
- Elution of the sample with an appropriate solvent.
An extensive review of available sorbents was carried out by Boyd et al.38 They found that the best recoveries in the literature were attributed to activated carbon-based sorbents. The high solubility and polarity of NDMA, however, make reproducibly adsorbing to activated carbon a challenge.42 An interesting study found that the key influencers of NDMA retention were micropore size, relative pore volume and surface chemistry.43 Specifically, an increase in hydrophobic sites and a reduction in oxygen-containing functional groups greatly increased NDMA retention. Astuti et al. also found that heat treatment at 800°C could be used to lower the surface oxygen content and thereby increase NDMA retention by 100% compared to a non-heated sorbent.44
SPE has several sources of NDMA contamination which may lead to false positives which anyone developing a method should be aware of. Firstly, activated carbon phases have been shown to catalyse formation of N-nitrosamines during extraction steps from secondary amines present in the sample.45 This effect has been shown to increase with prevalence of surface defects, carbonyl functionalities, surface area and access to dissolved/atmospheric oxygen.46 An exploration of alternative, but equally efficient, sorbent phases are needed to address this. Qian et al. made a vinyl/divinylbenzene polymer SPE sorbent cartridge capable of pre-concentrating N-nitrosamines for analysis at sub-ppt levels, which could be suitable if commercialised.47 The second cause of false positives relating to SPE is the presence of NDMA in the solvents or reagent waters used in the conditioning, washing, equilibration and elution steps. Parr and Joseph report finding NDMA in dichloromethane, which is the most used SPE extraction solvent for NDMA, as well as reagent waters.30 However, a recent study which looked at concentrations of N-nitrosamines in commercial and analytical grade solvents reported no NDMA detected in dichloromethane.48 This suggests that NDMA presence as an impurity will vary across manufacturers and highlights the importance of performing extraction blanks to understand baseline levels of NDMA for specific extraction conditions. One study even claimed contamination of NDMA from laboratory air,49 although this finding is unusual given that the compound is susceptible to photodegradation, and this has not been reproduced elsewhere.
SPE does have room to be made faster and more automated, which may result in better reproducibility. Tang et al. developed an online SPE method that utilised size exclusion chromatography to accurately quantify NDMA to sub-ppt levels in wastewater.50 The ability to automate procedures is highly valued in routine testing laboratories, so this method could potentially be adapted for drinking water analysis.
Solid-phase microextraction (SPME)
Solid-phase microextraction is a particularly useful technique for extracting volatiles from dirty or solid matrices. It is essentially a static headspace (HS) analysis, except sensitivity is increased by concentrating analytes on a fibre suspended in the sample headspace (Fig. 6). Compounds with an affinity for the fibre will bind to it during the extraction step. For quantitative analysis, the equilibration time, ratio of headspace to sample, extraction time, extraction temperature and fibre chemistry all need to be optimised.51
There have been a multitude of fibre types used for NDMA analysis but the most common coating phases have been a mixture of carboxen (CAR), divinylbenzene (DVB) or polydimethylsiloxane (PDMS).18,52–55 Llop et al. found that a 3-phase fibre made of DVB/CAR/PDMS recovered more NDMA than CAR/PDMS and polyethylene glycol (PEG).54 Importantly, they achieved an LOD of 5 ng/L with a fully automated method by optimising all aspects of the SPME, including time (60 min) pH (7), salt concentration (360 g/L), and temperature (45°C). Fan and Lin however, managed an even lower LOD of 0.5 ng/L for NDMA and an LOQ of 2.1 ng/L using a 2-phase CAR/PDMS fibre and high extraction temperature (85°C).55 They used a manual SPME technique, with a magnetic stir bar for agitation which should be able to be replicated on-line (automated) for future methods. NDMA is semi-volatile, so it makes sense that a higher extraction may be needed to increase recovery. However, this also has the potential of increasing noise from matrix adsorption to the fibre.
A current limitation of SPME is that it is designed for injection into hot injection ports on a gas chromatographic instrument only. There is, however, developing technology that would allow desorption into a liquid solution post fibre concentration, which could allow for liquid injection to a liquid chromatography system.51
SPME is preferred over static headspace injections for the increased sensitivity, although Zheng et al. found that using a single drop HS technique was 38 × more sensitive for NDMA analysis than an equivalent 1 mL solution.56 This technique shows promise, with few matrix effects compared to SPME, although it should be noted that the ratio of 1 mL liquid to 19 mL headspace used for this comparison is not an optimal equilibrium for sensitivity in HS analysis.
Other Extractions
Other NDMA extraction techniques have been reported in the literature for different water-based matrices. This includes Soxhlet extraction from sewage sludge,57 microwave assisted extraction, ultrasound assisted extraction,58 pressurised hot water extraction59 and trapping and concentrating on Tenax-TA liner for beer.60 This latter technique is of particular interest as purge and trap technology has advanced since the authors originally published their work. They reported an LOD of 10 ppt at the time, so this technique warrants revisiting with more modern liners for trapping.
A final note on extraction and pre-concentration is that care must be taken when trying to concentrate NDMA sample solutions by evaporation. NDMA is known to co-evaporate with solvents, so the evaporation step (if any) must also be optimised for high recoveries.35
Separation
To increase sensitivity, specificity and accuracy of measurement for NDMA, researchers often employ chromatographic techniques to separate the analyte of interest from others in the matrix. While the aforementioned extraction techniques do a good job of this, they are not specific enough to isolate NDMA completely. For this, we turn to chromatography.
Gas Chromatography (GC)
The most reported separation technique for NDMA analysis is gas chromatography (GC).30 Gas chromatography exploits the differences between compounds’ boiling points, vapour pressures, and polarities for separation from each another. NDMA is volatile enough to flow through a capillary column and so can take advantage of the superior resolution that can be achieved in gas phase separations without the need for lengthy derivatisation steps.23 Selection of the correct column chemistry (stationary phase) for retention and separation of the target compounds is crucial for sensitive and specific analysis.61 For NDMA, moderate to high polarity stationary phases are favoured.30
GC detectors for NDMA analysis to the required levels of sensitivity can be achieved with either chemiluminescence or mass spectrometry.30
Chemiluminescence has allowed for excellent LOQs (sub-ppt) and selectivity for NDMA in water and more complex matrices. This type of detection has been named in specific contexts as nitrogen chemiluminescence detection (NCD), thermal energy analyser (TEA) or nitrogen–phosphorus detection (NPD). The basic principle is that the labile nitrosyl group of NDMA becomes an NO radical when the compound is subjected to pyrolysis. NO is then oxidised to NO2- and this results in the emission of light in the near-infrared region, which is measured.62–65
Mass spectrometry (MS) is a technique which relies on applying a charge to molecules and then separating them based on their mass to charge ratio (m/z).59 A key step for mass spectrometry is the ionisation technique. Chemical ionisation (CI) is seen to be a softer technique than traditional electron impact ionisation (EI), allowing analytes to enter the quadrupole without fragmentation. According to a recent review by Parr and Joseph,30 both these techniques are still used extensively in the literature and GC–MS/MS (tandem mass spectrometry) with either ionisation technique is the preferred method for drinking water analysis. Note the authors found CI often reported greater levels of sensitivity than EI.30
Testing has been done to compare these techniques. Sanchez et al. tested the sensitivity for N-nitrosamines for three different detectors: flame ionisation detection (FID), NPD and MS and found MS to be the most sensitive, with similar precision across all three.64 Grebel and Suffet compared NCD, NPD and MS and found comparable sensitivity across all three detectors.65
Liquid Chromatography (LC)
Liquid chromatography exploits an analyte’s unique interactions with a selected liquid (mobile) phase and solid (stationary) phase. It has been used extensively for NDMA analysis, though is less widely used for water matrices than GC.66–70 Some N-nitrosamines are not volatile or thermally stable enough for analysis by GC, so researchers studying multiple N-nitrosamines at once tend to favour chromatography. The preferred column chemistry across these methods is reverse-phase C18, although the exact phase, gradients and mobile phases do differ once optimised.23,30,38,47,66–70
To achieve the required levels of sensitivity, only the mass spectrometer based detection method has been found to be suitable without derivatisation of NDMA.30 There is variability in the type of mass spectrometer, with triple quadrupole (MS/MS) being by far most common.23,30,38,47,68,69 The selectivity of running multiple reaction monitoring (MRM) methods on these instruments allow background noise to be minimised as much as possible, which is crucial when analysing very low molecular weight compounds. Briefly, MRM describes a method of selecting for an initial parent ion of the analyte of interest, then fragmenting this into smaller ions. One or more of these smaller “product” ions is selected for quantification.71
High-resolution MS detectors have also been very useful for situations where chromatographic resolution isn’t possible. False positives have been reported due to co-elution with N,N-dimethylformamide, which was investigated and resolved by use of high-resolution mass spectrometry.70 Using mass spectrometers of higher resolution allow compounds that appear to have the same parent and product ions in a typical triple quadrupole spectrometer to be distinguished from one another by using the accurate mass.
The method of ionisation for NDMA is important for minimising fragmentation of the compound. In-source fragmentation reduces sensitivity of MRM methods, so needs to be understood. Electrospray ionisation (ESI) and atmospheric pressure chemical ionisation (APCI) are both used extensively with no major influence on sensitivity or reproducibility.23,30,38,47,66–70 The presence of nitrogen groups in NDMA means that it will ionise better with positive charge mode than negative in the ionisation source.72
Fluorescence detection coupled to LC separation has also been reported for NDMA.73,74 These methods have excellent sensitivity, but require extra derivatisation steps that aren’t required in MS detection, as NDMA does not contain any fluorophores of its own.
Internal standards
This article has so far described an extensive range of extraction techniques for NDMA from water. As a check on recovery and detector response, many of these techniques utilise the stable isotope, D6-NDMA, as an internal standard. SPE techniques particularly have a variable recovery, so the use of an internal standard that has a behaviour closely representative of the analyte under extraction conditions is essential. D6-NDMA is readily available on the market as it is a requirement for EPA method 521.35
Conclusions
The aim of this review was to summarise techniques for the analysis of N-nitrosodimethylamine in drinking water. Note that the focus here was on highly sensitive laboratory-based methods to meet new regulatory limits. For a review of work on on-line sensors for monitoring of N-nitrosamines, the work of Beard and Swager is comprehensive.23
While the screening of drinking waters for NDMA may be new to New Zealand, it is not new for other countries. NZ analytical laboratories have a great existing body of work on which to develop sensitive and precise methods for the routine testing of NDMA and other N-nitrosamines.
The future may also hold requirements for testing of NDMA in other matrices. There has already been a focus on pharmaceuticals, but the food industry may be next with the European Food Safety Authority (EFSA) making a recommendation for standardisation of an N-nitrosamine analysis method in food matrices.10
Acknowledgements
Thank you to Grace McNicholl for drawing Fig. 6. Thanks to Justin Bendall and Paul Plieger for providing peer review. Thanks to Fonterra for sponsoring the work.
References
- Water Services (Drinking Water Standards for New Zealand) Regulations 2022. https://www.legislation.govt.nz/regulation/public/2022/0168/latest/whole.html (accessed 16/07/2023).
- Guidelines for drinking-water quality: fourth edition incorporating the first and second addenda. Geneva: World Health Organisation; 2022. License: CC BY-NC-SA 3.0 IGO.
- Kumar, M.; Shekhar, S.; Kumar, R.; Kumar, P.; Govarthanan, M.; Chamind, T. Environ. Pollut. 2023, 320, 121009.
- Tuesuwan, B.; Vongsutilers, V . J. Pharm. Sci. 2023, 112 (5), 1192-1209.
- Akkaraju, H.; Tatia, R.; Mane, S. M.; Khade, A. B.; Dengale, S. J. Regul. Toxicol. Pharm. 2023, 139, 105355.
- Fiddler, W.; Pensabene, J. W.; Doerr, R. C.; Dooley, C. J. Food Cosmet. Toxicol. 1977, 15 (5), 441-443.
- Muianga, C.; Roney, N.; Carlson-Lynch, H.; Citra, M.; Przybyla, J.; Heit, C. Toxicological profile for N-nitrosodimethylamine (NDMA). Registry, US Agency for Toxic Substances and Disease, Ed.; US Agency for Toxic Substances and Disease Registry website, 2023. https://wwwn.cdc.gov/TSP/substances/ToxSubstance.aspx?toxid=173
- United States Environmental Protection Agency. IRIS assessment for N-nitrosodimethylamine 1987. https://iris.epa.gov/ChemicalLanding/&substance_nmbr=45
- Šulc, M.; Hodek, P.; Stiborová, M. Gen. Physiol. Biophys. 2010, 29 (2), 175–185.
- Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J. K.; Del Mazo, J.; Hogstrand, C.; (Ron) Hoogenboom, L.; Leblanc, J.; Nebbia, C. S.; Nielsen, E.; Ntzani, E.; Petersen, A.; Sand, S.; Schwerdtle, T.; Vleminckx, C.; Wallace, H.; Romualdo, B.; Fortes, C.; Hecht, S.; Iammarino, M.; Mosbach‐Schulz, O.; Riolo, F.; Christodoulidou, A.; Grasl‐Kraupp, B. EFSA J. 2023, 21 (3), 7884.
- Tricker, A. R.; Spiegelhalder, B; Preussmann, R. Cancer Surv. 1989, 8 (2), 251-272.
- Hanst, P. L.; Spence, J. W.; Miller, M. Environ. Sci. Technol. 1977, 11, 403−405.
- Tuazon, E. C.; Carter, W. P. L.; Atkinson, R.; Winer, A. M.; Pitts, J. N. Environ. Sci. Technol. 1984, 18, 49−54.
- Plumlee, M. H.; Reinhard, M. Environ. Sci. Technol. 2007, 41, 6170−6176.
- Asami, M.; Oya, M.; Koji, K. Sci. Total Environ. 2009, 407 (11), 3540-3545.
- Cai, H.; Shen, C.; Xu, H.; Qian, H.; Pei, S.; Cai, P.; Song, J.; Zhang, Y. Sci. Total Environ. 2023, 857, 159530.
- Charrois, J.W.A. Boyd. J.M.; Froese, K.L.; Hrudey, S.E. J. Environ. Eng. Sci. 2007, 6 (1), 103-114.
- Chaves, R.S.; Rodrigues, J.E.; Santos, M.M. Benoliel, M.J. Cardoso, V.V. Anal. Meth. 2022, 14 (47), 4967-4976.
- Luo, Q.; Miao, Y.; Liu, C.; Bei, E.; Zhang, J.-F.; Zhang, L.-H.; Deng, Y.-L.; Qiu, Y.; Lu, W.-Q.; Wright, J. M.; et al. Chemosphere 2023, 315, ISSN 0045-6535.
- Mhlongo, S.H.; Mamba, B.B.; Krause, R.W. Phys. Chem. Earth A/B/C 2009, 34 (13–16), 819-824.
- Vizioli, B.D.C.; Hantao, L.W.; Montagner, C.C. Env. Sci. Poll. Res. 2021, 28 (25), 32823–32830.
- Liew, D.; Culbert, J.; Linge, K.; Farré, M. J.; Knight, N.; Morran, J.; Halliwell, D.; Newcombe, G.; Charrois, J. W. A. National occurrence of N-nitrosodimethylamine (NDMA). In: Recent Advances in Disinfection By-Products, Karanfil, T., Mitch, B., Xie, Y., Westerhoff, P. Eds.; ACS Symposium Series 2015, 1190, 135-149
- Beard, J. C.; Swager, T. M. J. Org. Chem. 2021, 86 (3), 2037-2057.
- Sharma, K. V . Sep. Purif. Technol. 2012, 88, 1-10.
- Sgroi, M.; Vagliasindi, F. G. A.; Snyder, S. A.; Roccaro, P. Chemosphere 2018, 191, 685-703.
- Schreiber, I. M.; Mitch, W. A . Environ. Sci. Technol. 2006, 40, 6007-6014.
- Choi, J.; Valentine, R . Environ. Sci. Technol. 2003, 37, 4871-4876.
- Bolduan, F.; Jodi, H. J.; Loewenschuss, A . J. Chem. Phys. 1984, 80 (5), 1739-1743.
- Andrzejewski, P.; Kasprzyk-Hordern, B.; Nawrocki, J. Environ. Sci. Technol. 2008, 42, 863-870.
- Parr, M. K.; Joesph, J. F. J. Pharm. Biomed. Analy. 2019, 164, 536 - 549.
- Malihi, F.; Wang, T . J. Chrom. Open 2022, 2, ISSN 2772-3917.
- Alsolais, A. M.; Althafar, Z. M.; Hasan, M. R.; Hakami, M. A.; Omar, B. I. A.; Hessien, K. B. G.; Alkhorayef, N.; Khan, F. R . J. Pharm. Neg. Results 2023, 14 (2), 2031–2043.
- Yin, M.; Hu, Y.; Fan, H.; Wang, Q.; Wang, M.; Wang, W.; Shi, C . J. Sep. Sci. 2022, 46 (5), e2200225.
- Fritzshe, M.; Blom, G.; Keitel, J.; Goettsche, A.; Seegel, M.; Leicht, S.; Guessregen, B.; Hickert, S.; Reifenberg, P.; Cimelli, A.; Baranowski, R.; Desmartin, E.; Barrau, E.; Harrison, M.; Bristow, T.; O’Neill, N.; Kirsch, A.; Krueger, P.; Saal, C.; Mouton, B.; Schlingemann, J. Eur. J. Pharm. Sci. 2022, 168, 106026.
- Munch, J. W.; Bassett, M. V. Method 521: Determination of Nitrosamines in Drinking Water by Solid Phase Extraction and Capillary Column Gas Chromatography with Large Vol. Injection and Chemical Ionization Tandem Mass Spectrometry (MS/MS); U.S. Environmental Protection Agency, 2004.
- Bamford, C. H. J. Chem. Soc. 1939, 12−17.
- Singer, G. M.; Taylor, H. W.; Lijinsky, W. Chem. Biol. Interact. 1977, 19 (2), 133-142.
- Boyd, J. M.; Hrudey, S. E.; Richardson, S. D.; Li, X. F. Trends Anal. Chem. 2011, 30 (9), 1410-1421.
- Cheng, R. C.; Hwang, C. J.; Andrews-Tate, C.; Guo, Y. B.; Carr, S.; Suffet, I. H. J. Am. Water Works Assoc. 2006, 98, 82–96.
- Hu, R.; Zhang, L.; Yang, Z . Water Sci. Technol. 2008, 58 (1), 143-151.
- Biziuk, M . Polish J. Env. Stud. 2006, 15 (5), 677-690.
- Bian, Y.; Wang, C.; Zhu, G.; Ren, B.; Zhang, P.; Hursthouse, A. S. Env. Eng. Res. 2018, 24 (1), 1–16.
- Dai, X., Zou, L., Yan, Z., Millikan, M. J. Hazard. Mat. 2009, 168 (1), 51-56.
- Astuti, M. P.; Jasemizad, T.; Padhye, L. P. J. Sep. Sci. 2020, 44 (2), 618-627.
- Padhye L.; Wang P.; Karanfil T; Huang C. H. Environ. Sci. Technol. 2010, 44 (11), 4161-41688.
- Padhye L.; Hertzberg, B.; Yushin, G.; Huang, C. H. Environ. Sci. Technol. 2011, 45, 8368-8376.
- Qian, Y.; Wu, M.; Wang, W.; Chen, B.; Zheng, H.; Krasner, S.W.; Hrudey, S.E.; Li, X.-F. Anal. Chem. 2015, 87, 1330–1336.
- Kosuri, E. R.; Bhanti, M.; Jaywant, M. A.; Han, M.; Wang, X.; Obeng, M. J. Pharm. Sci. 2023, 112 (5), 1225–1230.
- Kodamatani, H.; Sugihara, K.; Tanisue, T.; Kanzaki, Ryo.; Tomiyasu, T. Anal. Sci. 2020, 36, 1393-1399.
- Tang, H.; Li, Z.; Chen, H.; Xu, Y.; Jiang, X.; Du, E.; Lyu, Z.; Zheng, L.; Peng, M. Water 2022, 14, 2371.
- Schmidt, K.; Podmore, I. J. Mol. Biomarkers Diag. 2015, 06 (06).
- Grebel, J. E.; Young, C. C.; Suffet, I. H. Solid-phase microextraction of N-nitrosamines. J. Chrom. A 2006, 1117 (1), 11-18.
- Hung, H. W.; Lin, T. F.; Chiu, C. H.; Chang, Y. C.; Hsieh, T. Y . Water, Air, Soil Pollut. 2010, 213, 459-469.
- Llop, A,; Borrull, F.; Pocurull, E. J. Sep. Sci. 2010, 33 (23-24), 3692-3700.
- Fan, C. C.; Lin, T. S. Chemosphere 2018, 200, 48-56.
- Zheng, J.; Kirkpatrick, C. L.; Lee, D.; Han, X.; Martinez, A. I.; Gallagher, K.; Evans, R. K.; Mudur, S., V.; Liang, X.; Drake, J.; Buhler, L., A.; Mowery, M. D. AAPS J. 2022, 24 (23).
- Chen, W. H.; Wang, C. Y.; Huang, T. H. Chemosphere 2016, 161, 546-554.
- Jurado-Sanchez, B.; Ballesteros, E.; Gellego, M. Sci. Total Environ. 2013, 463-464, 293-301.
- Llop, A.; Borrull, F.; Pocurull, E. Talanta 2012, 88, 284-289.
- Billedeau, S. M.; Miller, B. J.; Thompson Jr, H. C. J. Food Sci. 1988, 53 (6), 1696-1698.
- Al-Bukhati, W. Q.; Noman, A.; Qasim, A. S.; Al-Farga, A. Int. J. Agri. Innov. Res. 2017, 6, 123-128.
- Firooz, D.H.F.; Lieb, R. D.; Rounbehler, D. P. Anal. Chem 1975, 47 (7), 1188-1191.
- Jurado-Sánchez, B.; Ballesteros, E.; Gallego, M. J. Sep. Sci. 2010, 33 (4-5), 610–616.
- Sanchez, B. J.; Ballesteros, E.; Gallego, M. J. Chrom. A 2007, 1154 (1-2), 66-73.
- Grebel, J. E; Suffet, I. H. J. Chrom. A 2007, 1175, 141-144.
- Huy, N. V.; Murakami, M.; Sakai, H.; Oguma, K.; Kosaka, K.; Asami, M.; Takizawa, S. Water Res. 2011, 45 (11), 3369-3377.
- Wang, W.; Hu, J.; Yu, J.; Yang, M. J. Environ. Sci. 2010, 22 (10), 1508–1512.
- Wang, W.; Yu, J.; An, W.; Yang, M. Sci. Total Environ. 2016, 551-552, 489–495.
- Topuz, E.; Aydin, E.; Pehlivanoglu-Mantas, E. Water, Air, Soil, Pollut. 2012, 223, 5793-5802.
- Yang, J.; Marzan, T. A.; Ye, W.; Sommers, C. D.; Rodriguez, J. D.; Keire, D. A. AAPS J. 2020, 22 (4).
- Ho, C. S.; Lam, C. W. K; Chan, M. H. M.; Cheung, R. C K.; Law, L. K.: Lit, L. C. W.; Ng, K. F.; Suen, M. W. M.; Tai, H. L. Clin. Biochem. Rev. 2003, 24, 3-12.
- Kadami, Y.; Favier, L.; Soutrel, I.; Lemasle, M.; Wolbert, D. Central Eur. J. Chem. 2014, 12 (9), 928-936.
- Cha, W.; Fox, P.; Nalinakumari, B. Anal. Chim. Acta 2006, 566, 109-116.
- Cárdenes, L.; Ayala, J. H.; González, V.; Afonso, A. M. J. Chrom. A 2002, 946 (1-2), 133-140.



