You may have heard the exciting news that work on sheep in New Zealand has led to US Federal Drug agency (FDA) sanctioned clinical trials to treat the fatal inherited neurodegenerative disease in children, CLN5 Batten disease (CLN5 neuronal ceroid lipofuscinosis, NCL). For me this has been an exciting journey from analytical chemistry to clinical neuroscience.
It began in 1980 when I accepted a position as a Research Officer working with Professor Bob Jolly in the Department of Veterinary Pathology and Public Health at Massey University, Palmerton North. Bob had established a considerable international reputation from his diagnosis of α-mannosidosis in cattle in collaboration with Dennis Hocking via biomolecular analysis of α-mannose rich oligosaccharides in blood and urine and subsequent assays of α-mannosidase activities. He had realised the importance of lysosomal storage diseases in domestic animals as models of human disease.
Encouraged by people including Nobel laureate Christian de Duve, he applied for NIH funding to pursue these studies highlighting their relevance to the emerging cognisance of human lysosomal storage diseases. While visiting a farm to collect some cow urine, his attention was drawn to a couple of blind South Hampshire sheep that also behaved like pet lambs. After initially dismissing them, he decided that a professional veterinary pathologist was obliged to investigate this thoroughly, had a post-mortem done, immediately noted the atrophied brain and diagnosed a ceroid lipofuscinosis, the diagnosis alerted from the similar brain atrophy in a pup he had diagnosed earlier.1 He procured some of the parental stock and established a research flock of these sheep to study as a model of the human disease.
My induction
The NIH provided the funds and I was employed to find out what was going on. I had done a degree in chemistry at Canterbury but was always attracted to biological chemistry and “doing something useful.” Biomedical research had been my career choice since my appetite was whetted by a short stint with Professor Robin Carrell in Christchurch Hospital in 1970, after which I went to the University of Toronto and then to University College London to develop my biological chemistry and biochemistry knowledge for this. By this time, 1980, and back in New Zealand, I was looking for something other than murdering healthy rats in the interests of human health and physiology, so the opportunity to study unhealthy animals to help human medicine was highly attractive.
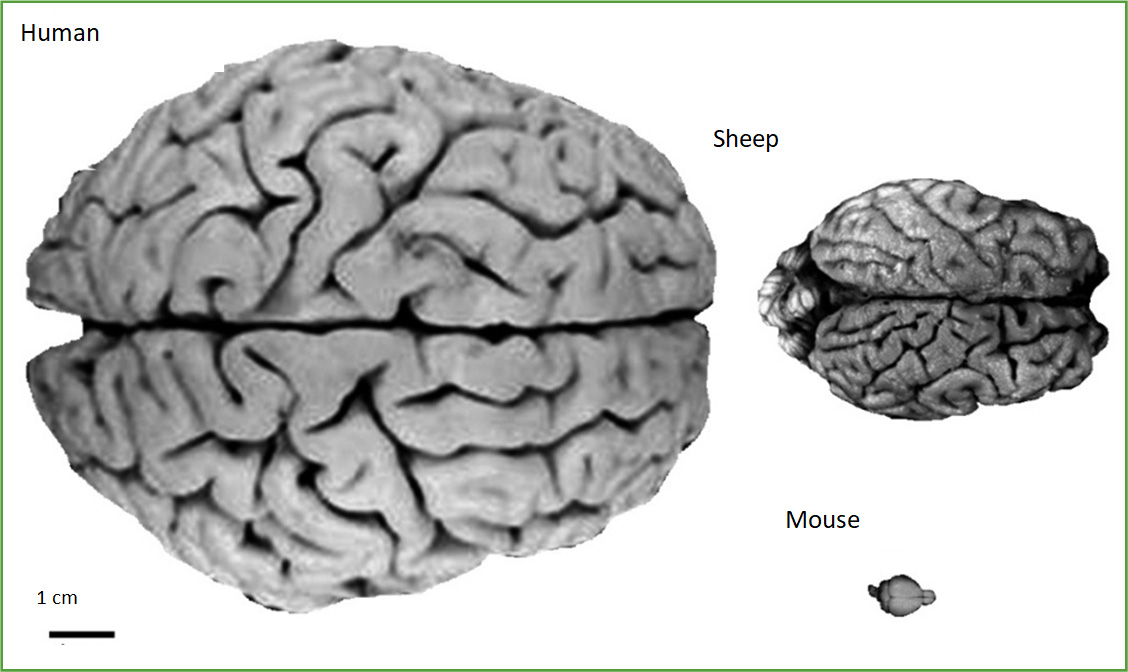
Sheep are ideal models. They are independent easy-care animals, particularly in our pastoral conditions, their gyrencephalic ovine brain is similar in size and physical organisation to non-human primates (unlike the tiny sulci and gyri-lacking smooth rat or mouse brain) and overall they are similar in size and organisation to human brains (Fig. 1). These facts have stimulated this longitudinal investigation into the ovine disease progression, and therapeutic efficacy and translatability of viral-mediated gene transfer in established CLN5 and CLN6 NCL sheep models.
The neuronal ceroid lipofuscinoses and lipid peroxidation
These are a group of inherited neurodegenerative lysosomal storage diseases of humans and other animals. Affected children start life normally but develop dementia, blindness and seizures, culminating in premature death. Characteristic pathological features are brain atrophy, retinal degeneration and the accumulation of fluorescent storage bodies in neurons and most other cells. Cases are relatively common and an estimate of 1 in 12,500 live births worldwide is broadly in line with the incidence in New Zealand. With intensive supportive therapy patients live for a number of years at great human and economic cost.
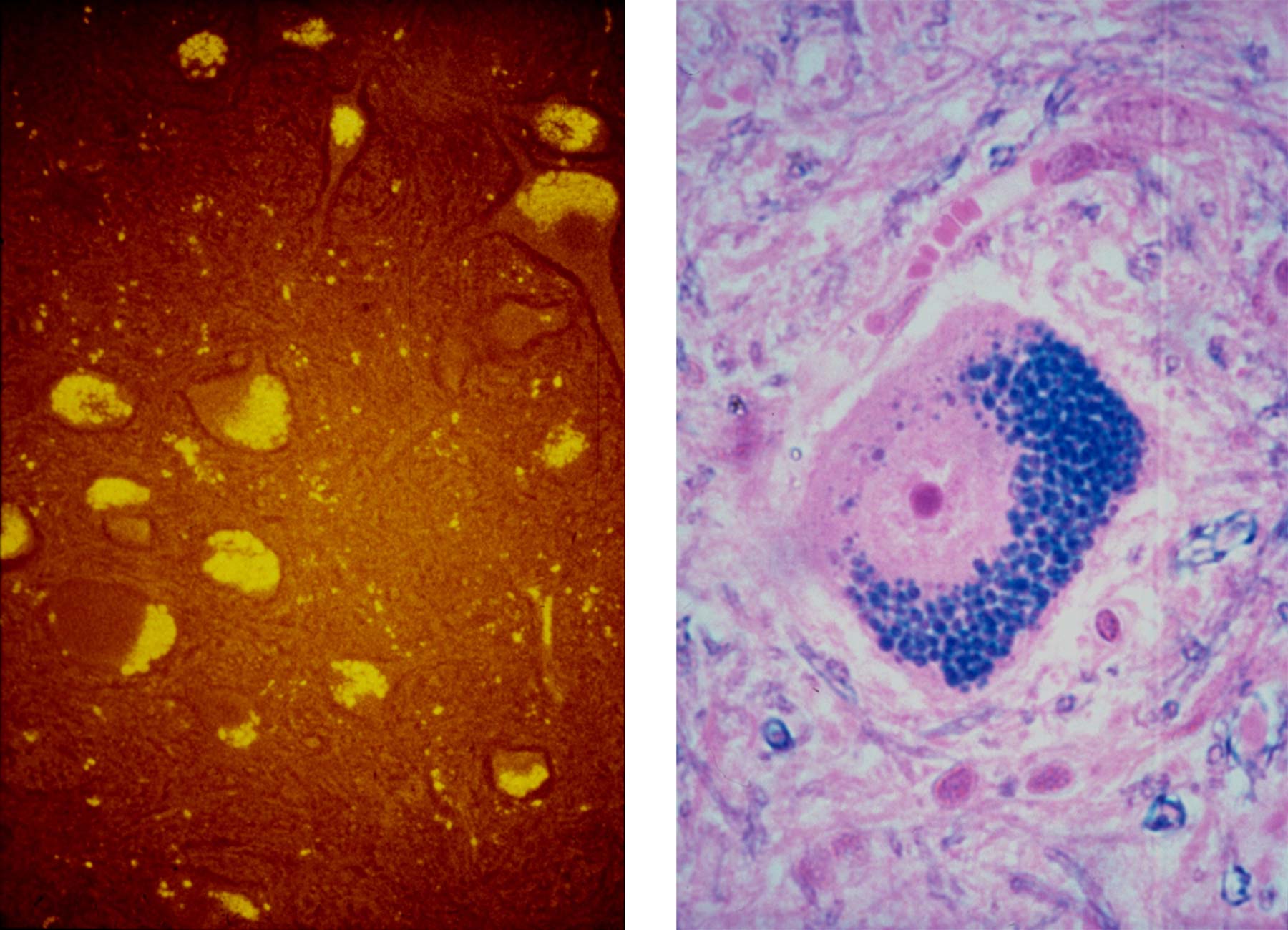
The name ceroid lipofuscinosis derives from the similar histochemical and fluorescent properties of the disease characteristic storage bodies and those of the intracellular dense body inclusions, ceroid and lipofuscin, still commonly thought to result from abnormal lipid peroxidation (Fig. 2). At that time the dogma was that these diseases arose from a failure of antioxidant protection causing a surfeit of free radical damage leading to peroxidation of lipids; the consequent aldehydes and ketones then cross linking to amines (in a pseudo aldol condensation) to form fluorescent Schiff base polymers resulting in the accumulation of “autofluorescent” lipofuscin storage bodies.
These characteristic intracellular inclusions that give the diseases their name are highly fluorescent in situ and were likened to the so called normal “age pigment” lipofuscin. Based on experiments by Chio and Tappel who had stewed up a variety of biological molecules with oxygen and got fluorescent polymers with spectra similar to lipofuscin,2 the same cause was postulated - abnormal lipid peroxidation products linking with amines. There were a number of publications attesting to a disease association with the abnormal peroxidation of polyunsaturated fatty acids, enhanced peroxidase activities, lack of antioxidant activity and so on, however the methodology in them was poor and the evidence only circumstantial at best. I thought these arguments of structural determination by analogy from similar fluorescence rather tenuous and it was not even clear where this abnormal peroxidation was meant to occur - in lysosomes, in the cell lumen or extracellularly.
Because of the rumen reduction of polyunsaturated fatty acids, ruminants have a very limited effective essential fatty acid supply and have special mechanisms to conserve them, thus a drain on them would be expected to lead to a net loss of polyunsaturated fatty acids. However, a careful analysis of the fatty acids of affected sheep phospholipids did not reveal any loss of peroxidation susceptible polyunsaturated fatty acids, which would be expected if the peroxidation mechanism was true.3 There is an adage in lysosomal storage diseases that if you figure out the structure of the stored compound you can guess the enzyme that should be next in the catabolic pathway, e.g. and re previously, the storage of oligosaccharides ending in an α-linked mannose indicated an α-mannosidosis, thus our next set of investigations centred on isolating the storage bodies and analysing their molecular composition. I’d done time in London isolating functional microsomes by gradient centrifugation and started off isolating storage bodies this way, but experience showed they behaved more like viral particles than microsomes and were refractory to sonication, which made it easy to obtain clean preparations.
Analytical chemistry
Given the predisposition to a lipidosis arising from the peroxidation hypothesis, the apparent histological lipoid nature of the storage bodies and an alternative hypothesis then fashionable in the literature - that the disease was caused by underlying defects in isoprenoid metabolism resulting in the accumulation of dolichols and/or derivatives - we started with a Folch extraction of storage body lipids and analysed them. Nearly all of the storage bodies dissolved in chloroform/methanol with ammonium acetate, and later we found in 100% formic acid with mercaptoethanol and also in lithium dodecyl sulphate or sodium dodecyl sulfate with heaps of mercaptoethanol.
This exercise in analytical chemistry established that the lipid content of the storage bodies was that expected of lysosome derived organelles and there was no sign of a loss of polyunsaturated fats to peroxidation. There was no fast migrating fluorescent material on silica gel thin layer chromatographs (TLC) that could equate to the postulated “polymalonaldehyde” either.4 Having samples around in air, or leaving TLC plates out in air overnight resulted in the creation of lots of fluorophores, so I have no doubt that it is easy to make fluorescent artefacts. In fact, preparative TLC showed that the lysosomal phospholipid, bis(monoacylglycero)phosphate, present in large amounts, had very high proportions of the essential polyunsaturated fatty acids linolenic and linoleic acids. No fluorophor was found, nor evidence for the highly fluorescent peroxidation formed polymer postulated to be the consequence of the peroxidation.5
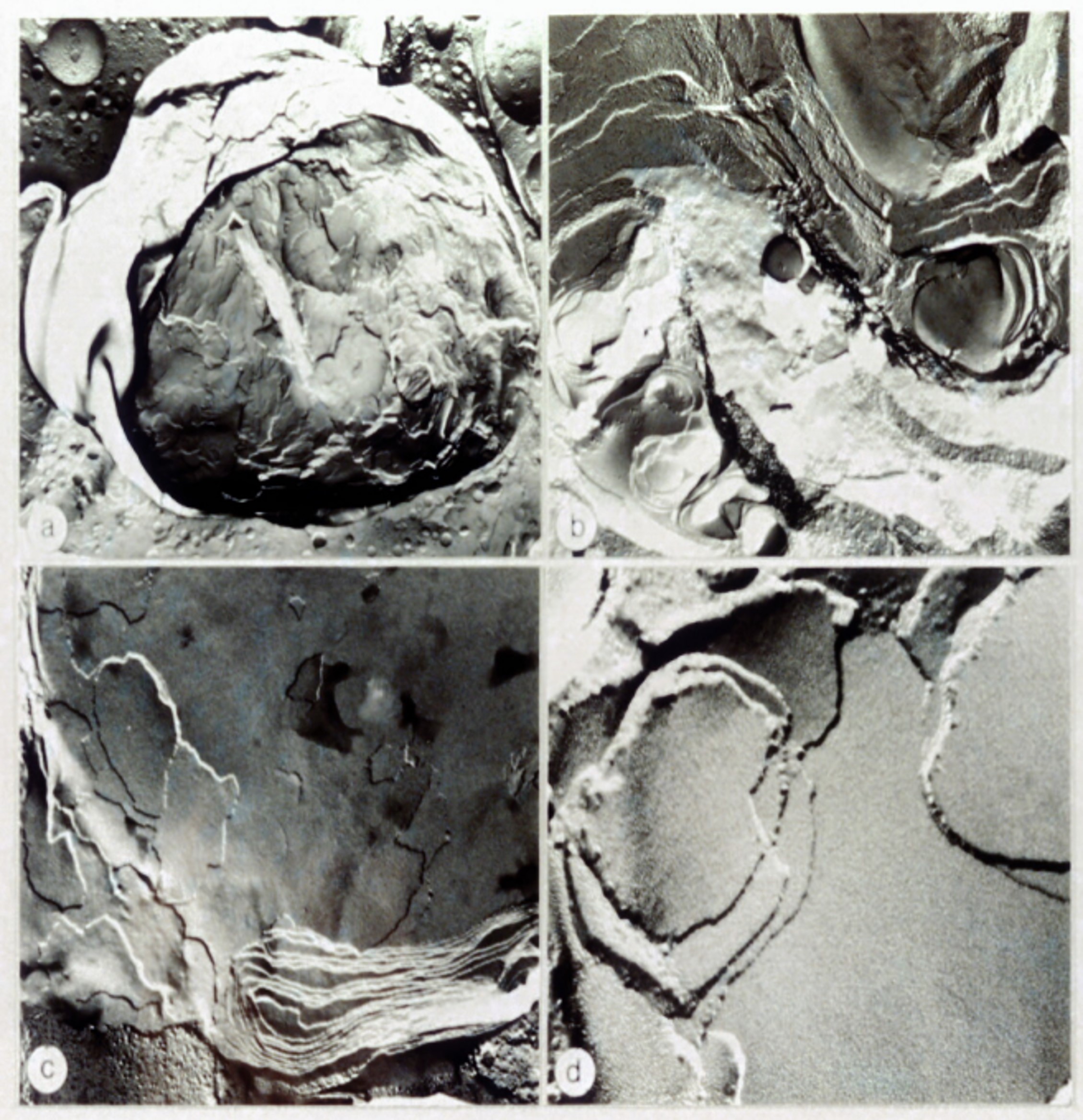
An efficient normal-phase HPLC method of analysing dolichols, cholesterol and ubiquinones was developed.6 These compounds were present in storage bodies only in amounts expected in lysosome derived organelles. However, their concentrations were lower than expected if the storage bodies were merely the aggregation of lysosomal membranes which indicated the accumulation something else. Freeze fracture and x-ray diffraction experiments showed that the apparently membranous nature of storage bodies disguised their solid nature (Fig. 3).7-8 Their isopycnic density (1.18-1.25) was far higher than expected of a lipid conglomerate. When in suspension or in situ the storage bodies were fluorescent, but when in solution the fluorescence disappeared and there was no significant fluorescent component, nor even one with a suitable uv/vis absorption spectrum.9
Analyses revealed that most of the storage body mass precipitated from the aqueous methanol/chloroform interface in Folch extractions. This insoluble residue was not fluorescent when directly irradiated with 366 nm light. Elemental analysis was consistent with it being proteinaceous; 53.19% C, 7.81% H, 12.71% N and 2.59% S by weight. Gravimetric analysis showed it to be about 70% of the total lipopigment mass, and Lowry protein estimations of sonicated suspensions of storage bodies gave the same proportion as protein.5
On the other hand, leaving lipid samples around in air, or leaving TLC plates out in air overnight resulted in the creation of lots of fluorophores, so I have no doubt that it is easy to make fluorescent artefacts. In fact, preparative TLC showed that the lysosomal phospholipid, bis(monoacylglycero)phosphate, present in large amounts, had very high proportions of the essential polyunsaturated fatty acids linolenic and linoleic acids. No fluorophor was found, nor evidence for the highly fluorescent peroxidation formed polymer postulated to be the consequence of the peroxidation.5
An efficient normal-phase HPLC method of analysing dolichols, cholesterol and ubiquinones was developed.6 These compounds were present in storage bodies only in amounts expected in lysosome derived organelles. However, their concentrations were lower than expected if the storage bodies were merely the aggregation of lysosomal membranes which indicated the accumulation something else. Freeze fracture and x-ray diffraction experiments showed that the apparently membranous nature of storage bodies disguised their solid nature (Fig. 3).7-8 Their isopycnic density (1.18-1.25) was far higher than expected of a lipid conglomerate. When in suspension or in situ the storage bodies were fluorescent, but when in solution the fluorescence disappeared and there was no significant fluorescent component, nor even one with a suitable uv/vis absorption spectrum.9
Analyses revealed that most of the storage body mass precipitated from the aqueous methanol/chloroform interface in Folch extractions. This insoluble residue was not fluorescent when directly irradiated with 366 nm light. Elemental analysis was consistent with it being proteinaceous; 53.19% C, 7.81% H, 12.71% N and 2.59% S by weight. Gravimetric analysis showed it to be about 70% of the total lipopigment mass, and Lowry protein estimations of sonicated suspensions of storage bodies gave the same proportion as protein.5
When this residue was suspended as a dry powder in glycerol it was highly fluorescent in the fluorescence microscope, but so are glycerol suspensions of casein and bovine serum albumin, indicating that this apparent fluorescence did not arise from a classical fluorophore. The metal content of the isolated storage bodies was consistent with a lysosomal origin of them and did not support a metal catalysed peroxidative mechanism of lipopigment formation.10
We thought it likely the fluorescence arose from the environmentally dependent fluorescence of protein, in a similar way to the observed fluorescence of the globules of protein that accumulate in the livers of people with alpha‑1‑antitrypsin (alpha‑1‑protease inhibitor) deficiency.11 However, it was a rather unusual protein, being highly insoluble in most protein solvents and soluble in lipid solvents. With wonderful help from Gill Ellis (then Barns) we found that if you treated it really gently and ran it in LDS gels at low loading you could detect two Coomassie insensitive bands with a specially developed silver stain technique; a dominant band with an apparent molecular mass of 3,500 kDa and a lesser component with an apparent mass of 14,800 kDa, and some smears in between.12 I was excited and went to a number of conferences about these inclusions where I realised how entrenched the “dangerous free radical damage” dogma is. I also found that the evidence these people presented for it was largely cooked up, rather à la Chio and Tappel. The dogma around free radical damage continues to persist.
"The similarities between lipofuscin and the storge bodies in ceroid-lipofuscinosis suggest a similar composition, albeit for different reasons to those usually advanced."
Dense bodies have also been observed in neurons of rat brains into which protease inhibitor leupeptin had been perfused.13 They formed quickly, their formation was reversible, and they had structural and fluorescent properties that drew attention to their similarity to lipofuscin and the storage bodies in the ceroid-lipofuscinoses. Lipofuscin and other similar intracellular inclusion bodies are also likely to be largely proteinaceous, and the consequence of general disruptions in lysosomal function particularly affecting lysosomal proteolysis.
After gazing at these two bands on gels and lots of unsuccessful efforts to separate them, it occurred to me when calculating mole % of each protein rather than the masses that the 3,500 kDa band was about 90% of the moles, so if it was one protein it should be sequencable without further purification. Without further ado and with the help of the able Julian Reid we loaded the protein into an ABI automatic N-terminal Edman degradation sequencer. The result was astonishing, a clear read of the 40 amino acids from one dominant protein, identical to the c-subunit of mitochondrial ATP synthase, and of the first 10 amino acids of another minor component, the c-subunit of vacuolar ATPase.14 Later work in collaboration with Sir John Walker and Ian Fearnley, then at the MRC Laboratory of Molecular Biology in Cambridge, established that the c-subunit of ATP synthase stored is the complete and normal 75 amino acid peptide, i.e. not truncated or covalently modified in any way except for the usual and completely normal trimethylation of a lysine.15 It was originally called the mitochondrial proteolipid as it is soluble in lipid solvents, having been discovered by precipitation from Foch extractions, is insoluble aqueous solutions and stains poorly with Coomassie blue.
Quantitative analyses, comparing the sequencer yield to the total amino acids loaded, indicated that this protein alone accounts for the bulk of the storage bodies’ mass. We subsequently found, via sequencing from gel bands that the 14,800 kDa band is an oligomer of subunit c. Whether the vacuolar subunit is a genuinely stored component or arises from the lysosomal membrane is not resolved but the variability of yield between different storage body preparations suggests the latter. We reconstituted these bodies from normal phospholipids and Folch extracted c-subunit and they were fluorescent. This led us to conclude that the fluorescence is an aggregate property, not an intrinsic molecular one.9
People and NCLs
Meanwhile progress was being made on NCL genetics. Originally the human diseases were classified into four forms; infantile, late infantile, juvenile and adult, based on the age of presentation of clinical symptoms and the clinical progress. An infantile form, INCL (CLN1), is most common in Finland. Two late infantile forms, classical and variant LINCL (CLN2 and CLN6) and a juvenile form, JNCL (CLN3), occur worldwide as do several other variants. A confusing number of eponyms have been used to describe these diseases. The best known, Batten disease, is usually used as a generic term for the group as a whole. Over time, 13 different genetically distinct forms have been recognised, now designated CLN 1-8 and 10-14, each gene having a range of disease causative mutations (www.ucl.ac.uk/ncl).
I was very interested in determining the relevance of our sheep findings to NCLs in humans and other animals. In a series of visits to New York, Montreal, Helsinki and particularly Cambridge, we established that the complete and normal ATP synthase c-subunit is also stored in the juvenile and late infantile forms.16 This was exciting. Subsequent studies showed that it is stored in all forms of Batten disease in humans and a large number in animals, with the exception of the infantile form, where we discovered specific storage of the sphingolipid activator proteins (SAPs or saposins), A and D.17 General protein turnover is not disturbed. This led to the concept that a number of gene products are necessary and unique components of a subunit c turnover pathway. However, there is nothing obvious in the gene products that interlink them. Some are soluble lysosomal proteins, some are not and there is increasing evidence of interactions between some of them in the autophagic pathway to lysosomal digestion. Along the way we found and established another flock, of Borderdale sheep with a CLN5 form, being a dysfunction of a soluble lysosomal protein.18
The nature of the international conferences was the other exciting thing. Initially they were very medically focused but progressed through to a considerable parent and support group involvement by 1990, led by the late Lance Johnston and Elaine, his wife. They built up the Batten Disease Support and Research Association (BDSRA) to provide educational material, beds, wheelchairs and furniture suitable for the patients and conferences brought all the parties together, including healthy siblings of affected children who have their own demons. Meeting parents brought me face-to-face with the human realities of the NCLs and gave me a sense of vocation. I had not fully appreciated the halo of psychological stress that affects a patient’s close family, siblings’ guilty anxiety at being normal but in fear of their fertility, and all this in addition to the patient’s need for 24 hour care with demands at any hour of the day or night that drains parents and caregivers. This is after the stress and loneliness of years of having no credible diagnosis for their children’s obvious decline.
Parents became friends and I became interested in joining them in communicating the full costs of the disease, both financial and emotional. In New Zealand we fall under the Australian branch of the BDSRA. They can be contacted at https://bdsraaustralia.org/contact. I have attended a number of their conferences found them informative and humbling. They were very interested in and supportive of our research and what it might mean for them. We had some meetings in New Zealand bringing together researchers, parents, carers, research funders and some patients too (Fig. 4).
Batten disease cases have been reported in Māori, Pacifica, Pakeha and New Zealanders of Indian descent.

Towards treatments
We continued with the pathology, particularly the neuropathology, and noticed early neuroimmune response and glial activation that might drive the disease.19-20 However, an experiment to treat sheep with chronic oral administration of anti-inflammatory minocycline did not prevent neuroinflammation or disease progression.21 We are still working on this, but experiments with gene therapy have taken over. These began at the instigation of Stephanie Hughes who had returned to New Zealand after a postdoc with Dr Beverley Davidson who encouraged this. This was the time of the anti-science anti-GM/GE furore which made progress difficult. The practical and administrative impediments invented were extremely onerous, not helped by a fretful approach of university administrators who were not all supportive. Fortunately Otago stepped in and gave Stephanie a position and a brief to make vectors.
The concept is really neat. To make a vector one starts with a virus known to be able to penetrate into cells and discharge genetic cargo that will use the host cell machinery to make more virus. All the genetic information coding for the replication of this virus is removed from the vector and is replaced by the code for the protein wanted, in this case a good copy of the dysfunctional gene. The result is a vector particle capable of entering a cell and expressing a healthy protein, but not of replicating vector or any viral proteins. Trillions of copies can be cloned, free of the risk of any of the native virus.
Vectors into sheep
Meanwhile the highly proficient and dedicated Nadia Mitchell had joined us and was doing an excellent job. In short order the lab was humming, we cloned the normal ovine CLN5 and CLN6 genes in collaboration with Tony Frugier and Imke Tammen in Sydney and determined the mutations in the sheep.18,22 Stephanie cloned these sequences into lentiviral and AAV9 vectors and we injected these into the brain parenchyma of affected sheep.23
CLN5-affected Borderdale sheep received lentiviral- or AAV9-mediated gene therapy between 2.1 - 3.5 months of age, prior to the onset of neurological disease signs. Viral vectors were delivered bilaterally into the cerebral lateral ventricles (intracerebroventricular; ICV) and to two intraparenchymal (IP) (cortical) sites which have been shown to degenerate significantly in the affected sheep. Clinical, cognitive and behavioural tests were begun, as were quantitative central nervous system imaging techniques, to assess disease progression and monitor therapeutic correction longitudinally.
The study endpoint was initially defined as 18 months of age, equivalent to an advanced disease state in untreated affected sheep, at which age the treated sheep were to be euthanised and collected tissues evaluated for any pathological amelioration. Once it became apparent that the treatment was functionally efficacious, the study was continued until five of the six treated sheep were euthanised at 27 months of age, exceeding the typical humane endpoint for untreated CLN5 deficient animals of 22 months of age. Their only symptoms were related to retinal degeneration and they became blind and clumsy, but post mortem examination revealed none of the parieto-occipital cortical atrophy typical of untreated affected sheep. The clinical outcomes were similar for both of the viral vector platforms tested.

Results from the CLN6 trials were not as spectacular, with only one of the six treated sheep responding positively. This was actually a pleasant surprise. Aside from genetic classification, the NCL gene products fall into two general subclasses – bona fide soluble glycosylated lysosomal proteins (CLN1, CLN2, CLN5 CLN10, CLN13) or predicted transmembrane proteins of unknown function. The lysosomal glycoproteins are taken up into lysosomes via a mannose 6 phosphate marker receptor, both intracellularly and extracellularly via the plasma membrane, and some of the protein produced is excreted from cells. This means that treated cells can cross-correct affected cells.
On the other hand, the dogma is that the insoluble membrane bound lysosomal proteins, of which CLN6 is one, are cell intrinsic, ruling out cross correction. However, an analysis of CLN6/normal chimeric sheep we created by embryo fusion (Fig. 5) indicated that normalised cells do correct affected cells, so long as their location and connectivity are appropriate.24
A number of improvements enhanced subsequent trials. We settled on recombinant adeno-associated viral vector serotype 9 (AAV9) as the vector backbone as this had become widely used worldwide and commercially available, allowing the testing of much higher titres and did not scare the horses as much as using lentiviral constructs. The vector does not integrate into the host genome, instead persisting in an extrachromosomal state as episomal concatemers in the host cell nucleus. In dividing cells the episomal DNA is not replicated with the host cell DNA and the AAV genome is lost at mitosis. However, in post-mitotic cells, such as differentiated neurons, AAV episomes remain intact providing sustained transgene expression and therapeutic protein production for the life of the host cell. Thus one set of injections into the right parts of the central nervous system should last indefinitely.
Gene Therapy in Sheep

A construct expressing enhanced green fluorescent protein was purchased from the vector core at the University of Pennsylvania. Nadia used this to map the consequences of different routes of administration. We settled on the intracerebroventricular injections alone (Fig. 6) as they bypassed the blood-brain barrier and led to widespread transduction of the central nervous system, notably parts of the brain susceptible to disease atrophy (Fig. 7). Higher doses were more efficient. The hysteria around GM/GE issues had abated and some of the more inane and onerous housing restrictions were relaxed so the sheep could now graze in the open, a massive improvement for them and for us. Intravitreal injections of vector added to the protocol prevented the remaining vision pathology subsequent to ICV injections alone.25
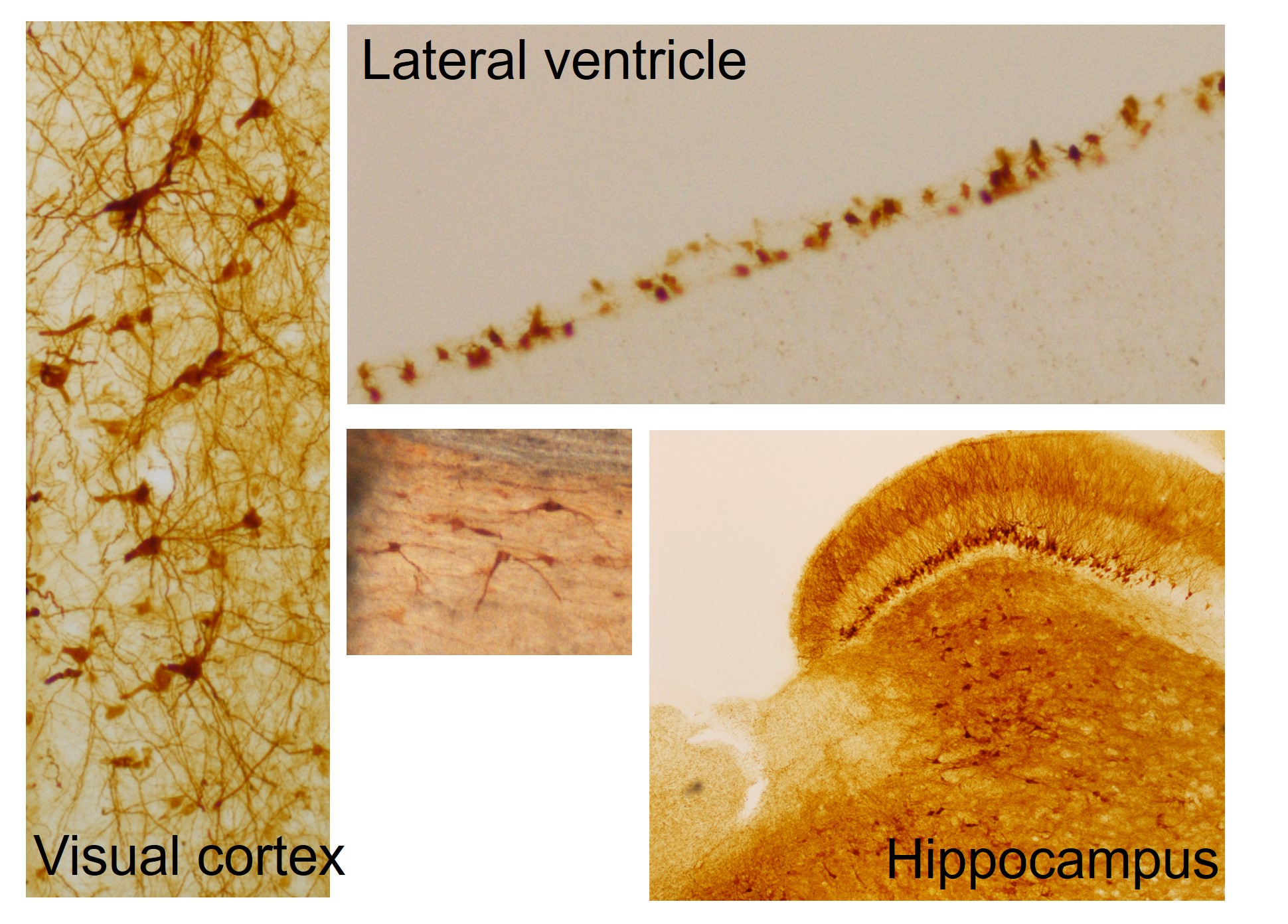
We also realised the benefits of long-term in vivo monitoring of treated sheep and the need to justify that in applications for translation of the gene therapy to human medicine. For this we developed tests that included a neuro-opthalmic examination assessing ocular function, the central visual pathways and the visual cortex.26 Auricular (acoustic startle) reflexes - the movement of the pinna in response to a loud noise - were also tested. Mentation, gait, head carriage and postural traits, as well as manifest tremor or seizure onset, were assessed while sheep were herded up a set of stairs and a graded slope to the testing facility. Functionality of the optic cranial nerve II, visual cortex and all cerebral areas associated with motor function were assessed by behavioural vision testing through negotiation of a maze in the field. Sheep were assigned a clinical score after examination by two independent assessors.
AAV mediated GFP
Commercial AAV9-GFP
AAV9 delivery in sheep
Intraventricular delivery results in good penetrance through the brain and spinal cord
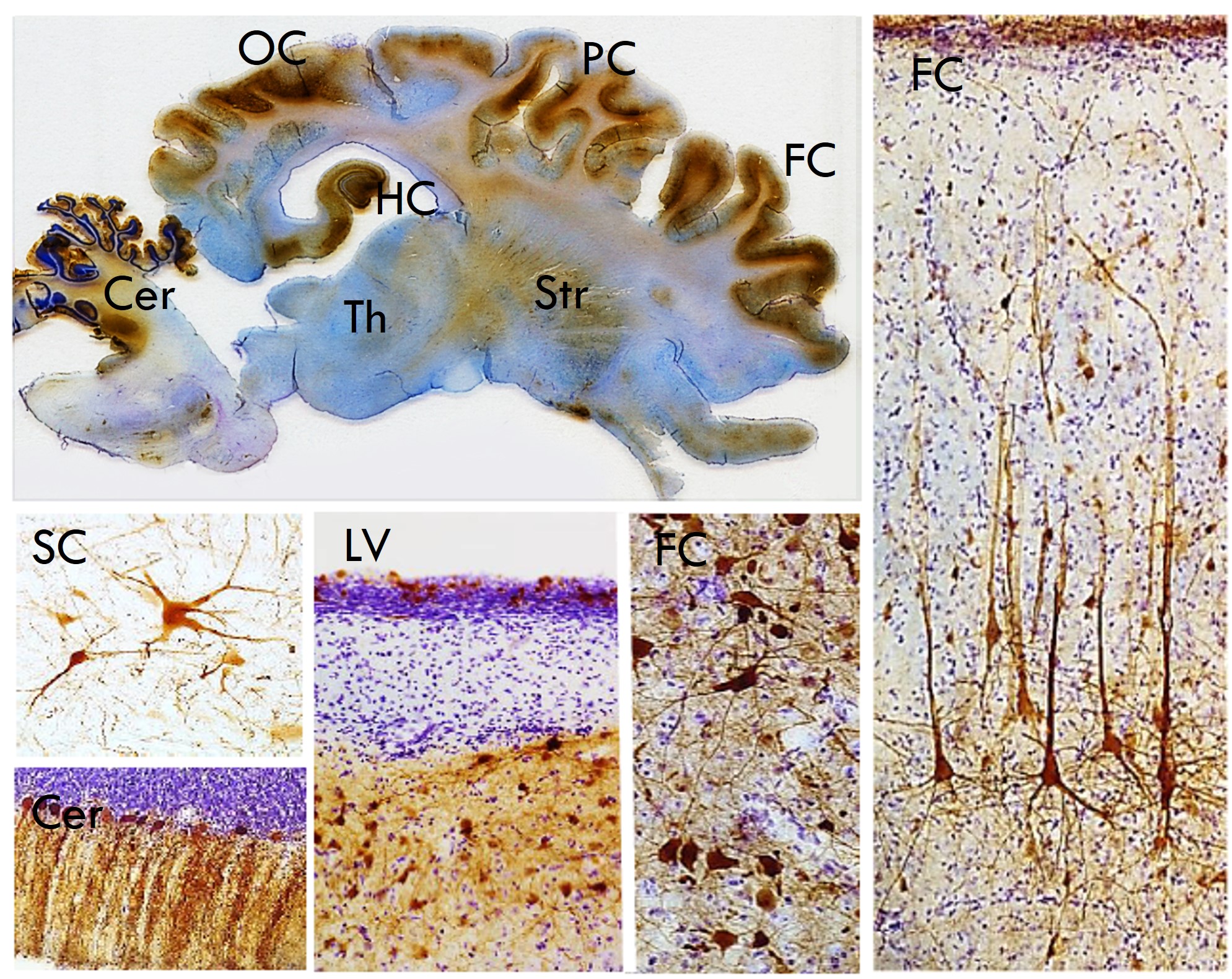
Computed tomography (CT) scans were performed every 2 months on animals who received intracranial gene therapy and intracranial volumes from the experimental animals were then compared with historical cohorts of untreated affected and heterozygous control sheep of both genotypes. Magnetic resonance imaging (MRI) was instigated after visiting some French colleagues and a longitudinal study of both CLN5 and CLN6 affected brains performed in collaboration with Dr Tracy Melzer, at the NZ Brain Research Institute.27-28 Ophthalmic examinations were performed on the treated sheep by an independent veterinary ophthalmologist. Electroretingraphic (ERG) and funduscopic techniques to monitor disease-associated loss of vision longitudinally were developed by Katharina Russell.29
The treated sheep were disease-free. They benefitted from a profound improvement in quality of life, preservation of cognitive and neurological function and normalisation of intracranial volume and brain structure compared to untreated animals. Longitudinal studies of non-invasive biomarkers were used to assess the efficacy of lentiviral and AAV-based therapeutics in sheep in vivo. Monthly neurological examination and clinical scoring using a sheep-specific rating scale revealed the long-lasting functional efficacy of treatment, with treated animals being clinically indistinguishable from untreated heterozygous controls until 20.5 ± 0.6 months and 24.1 ± 1.5 months for the lentiviral and AAV9 cohorts respectively, when they begin to show effects of their visual deficits. Aside from this loss of vision, at 27 months of age treated sheep were still highly interactive with their environment. Longitudinal monitoring showed that the CLN5 sheep did really well, likewise the CLN6 treated ones.
To clinical trials
While this was happening, we were trying to find an entity willing to give human trials a go, or at least work towards that, and had been to lots of conferences and meetings with parents to encourage that. The BDSRA was very supportive. Drug companies, wanting millions of sales, calculated that there was not enough money in it. Pharmac thought the disease was not serious enough as only a small number of people were afflicted, to my mind a very strange world view. Two American parents contacted us who were very interested in the CLN6 treatment. Their agent visited us, and we provided all the information we could and offered to help. However, they decided to do it all in America and our involvement ceased, somewhat to my chagrin and to the relief of our administration.
By now Nadia was in charge. The good news is that we were contacted by an American firm, Neurogene, who were really keen on giving human gene therapy of CLN5 a go. Their principal visited us, Nadia provided a really large and comprehensive summary of our work including results of our trials in sheep in the demanding format for US FDA approval.30 They found these data compelling and human clinical trials have begun. So far so good. We have also established sheep as an ideal model species and others follow.31,32
Unfinished business
This is not the end though. Nadia and co are setting up for long term studies of the treated sheep to see if any problems arise later, so we can preview any need that treated people may have. This includes the issue of non-central nervous system pathology that may become a problem later, and we still have no really satisfactory explanation of the fluorescence. A paper appeared in J. Phys. Chem. that Sir John Walker and I are interested in,33 and we are now in contact with the authors. BUT this still leaves the big unanswered question from the beginning. Why the specific storage of subunit c?
Acknowledgements
Firstly, thank you to all my colleagues and co-workers at Massey, Lincoln, Otago and overseas who have played integral roles in this adventure. Sorry I could not name you all, but you know who you are. Also thank you to the parents for their staunch support, good will and encouragement. It’s been a privilege to meet you all. None of this would have been possible without the financial support and encouragement from various charity funding bodies who have been there for us. We realise it is not intuitive that funding research on sheep is a way to help children’s health needs so thank you to the Neurological Foundation of New Zealand, CureKids New Zealand, The Canterbury Medical Research Foundation, Lysosomal Diseases New Zealand (LDNZ) and especially the Batten Disease Support and Research Association (BDSRA) and and US NIH.
References
1. Jolly, R. D.; West, D. M. NZ Vet. J. 1976, 24, 123.
2. Chio, K. S.; Reiss, U.; Fletcher, B.; Tappel, A.L. Science 1969, 166, 1535-1536.
3. Palmer, D.N.; Husbands, D.R.; Jolly, R.D. Biochim. Biophys. Acta 1985, 834, 159‑163.
4. Gutteridge, J.M.C.: Kerry, P.J.: Armstrong, D. Biochim. Biophys. Acta 1982, 711, 460‑465.
5. Palmer, D.N.; Husbands, D.R.; Winter, P.J.; Blunt, J.W.; Jolly, R.D. J. Biol. Chem. 1986, 261, 1766‑1772.
6. Palmer, D.N.; Anderson, M.A.; Jolly, R.D. Anal. Biochem. 1984, 140, 315‑319.
7. Palmer, D.N.; Martinus, R.D.; Barns, G.; Reeves, R.D. Jolly, R.D. Am. J. Med. Genet. 1988, Supp. 5, 141‑158.
8. Jolly, R.D.; Shimada, A.; Craig, A.S.; Kirkland, K.B.; Palmer, D.N. Am. J. Med. Genet. 1988, Supp. 5, 159‑170.
9. Palmer, D.N.; Bayliss, S.L.; Clifton P.A.; Grant, V.J. J. Inherit. Metab. Dis. 1993. 16: 292‑295.
10. DeLellis, R.A.; Balogh, K.; Merk, F.B.; Chirife, A.M. Arch. Path. 1972, 94, 308-316.
11. Palmer, D.N.; Barns, G.; Husbands, D.R.; Jolly, R.D. J. Biol. Chem. 1986, 261, 1773‑1777.
12. Ivy, G.O.; Schottler, F.; Wenzel, J.; Baudry, M.; lynch, G. Science 1984, 226, 985-987.
13. Palmer, D.N.; Martinus, R.D.; Cooper, S.M.; Midwinter, G.G.; Reid, J.C.; Jolly, R.D. J. Biol. Chem. 1989, 264, 5736‑5740.
14. Fearnley, I.M.; Walker, J.E.; Martinus, R.D.; Jolly, R.D.; Kirkland, K.B.; Shaw, G.J.; Palmer, D.N. Biochem. J. 1990, 268, 751‑758.
15. Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Am. J. Med. Genet. 1992, 42, 561-567.
16. Palmer, D.N.; Jolly, R.D.; van Mil, H.C.; Tyynelä, J.; Westlake, V.J. Neuropediatrics 1997, 28, 45-48.
17. Tyynelä, J.; Palmer, D.N.; Baumann, M.; Haltia, M. FEBS Lett. 1993, 330, 8-12.
18. Frugier, T.; Mitchell, N.L.; Tammen, I.; Houweling, P.J.; Arthur, D.G.; Kay, G.W.; van Diggelen, O.P.; Jolly R.D.; Palmer, D.N. Neuorbiol. Dis, 2008, 29, 306-315.
19. Oswald, M. J.; Palmer, D.N.; Kay, G.W.; Shemilt, S.J.A.; Rezaie, P.; Cooper, J.D. Neuorbiol. Dis, 2005, 20, 49-63
20. Kay, G. W.; Palmer, D.N.; Rezaie, P.; Cooper, J.D. Brain Pathol. 2006, 16, 110-116.
21. Kay, G.W.; Palmer D.N. 2013. J. Neuroinflammation 2013, 10:97.
22. Tammen, I.; Houweling, P.J.; Frugier, T.; Mitchell, N.L.; Kay, G.W.; Cavanagh, J.A.L.; Cook, R.W.; Raadsma, H.W.; Palmer, D.N. Biochim. Biophys. Acta 2006, 1762, 898-905.
23. Linterman, K.S.; Palmer, D.N.; Kay, G.W.; Barry, L.A.; Mitchell, N.L.; McFarlane, R.G.; Black, M.A.; Sands, M.S.; Hughes, S.M. Hum. Gene Ther. 2011, 22, 1011-1020.
24. Barry, L.A.; Kay, G.W.; Mitchell, N.L.; Murray, S.J.; Jay, N.P.; Palmer, D.N. PLoS ONE 2022, 17(4): e0261544. https://doi.org/10.1371/journal. pone.0261544
25. Murray, S.J.; Russell, K.N.; Melzer, T.R.; Gray, S.J.; Heap, S.J.; Palmer, D.N.; Mitchell, N.L. Exp. Eye Res. 2021, doi: https://doi.org/10.1016/ j.exer.2021.108600.
26. Russell, K. N.; Mitchell, N. L.; Wellby, M.P.; Barrell, G.K.; Palmer, D.N. Data Brief 2021, 37, 107188 doi.org/10.101016/j.dib2021.107188
27. Ella, A.; Barrière, D.; Adriaenssen, H.; Palmer, D.N.; Melzer, T.R.; Mitchell, N.L.; Keller, K. Anim. Front. 2019, 9, 44-51. doi.org/10.1093/af/vfz024
28. Murray, S.J.; Almuqbel, M.A.; Felton, S.A.; Palmer, N.J.; Myall, D. J.; Shoorangiz, R.; Ella, A,; Keller, M.; Palmer, D.N.; Melzer, T.R.; Mitchell N.L. Brain Commun. 2022, (In Press)
29. Russell, K.N.; Mitchell, N.L.; Anderson, N.G.; Bunt, C.R; Wellby, M.P., Melzer, T.R.; Barrell G.K.; Palmer, D.N. Brain Behav. 2018 8e01096. doi:10.1002/brb3.1096
30. Mitchell, N.L.; Russell, K.N.; Wellby, M.P.; Wicky, H.E.; Barrell, G.K.; Melzer, T.R.; Gray, S.J.; Hughes, S.M.; Palmer, D.N. Mol. Ther. 2018, 2366-2377. https://doi.org/10.1016/j.ymthe.2018.07.015
31. Eaton, S.L.; Murdoch. F.; Rzechorzek, N.M.; Thompson, G.; Hartley, C.; Blacklock, B.T.; Proudfoot, C.; Lillico S.G.; Tennant, P.; Ritchie, A.; Nixon, J.; Brennan, P.M.; Guido, S.; Mitchell, N.L.; Palmer, D.N.; Whitelaw, B.A., Cooper, J.D.; Wishart, T.M. Cells 2022, 11, 2641. https://doi.org/10.3390/cells11172641
32. O’Leary, C.; Fortea, G.; Mitchell, N.; Youshania, A.S.; Wellby M.; Russell, K.; Palmer, D.; Henckaerts, E.; Asl, I.K.; Bigger, B.. Mol. Genet. Metab. 2019, 126, S17–S156.
33. Morzan, U.N.; Mirón, G.G.; Grisanti, L.; González, M.C.; Lebrero, G.S.; Schierle, K.; Hassanali, A. J. Phys. Chem. B 2022, 126, 7203−7211.