


The click chemistry concept
I confess that, after last year’s initial slight disappointment with the outcome, this year I did not stay up and watch the Chemistry Nobel prize announcement live on YouTube1 on the night of 5th October 2022. Of course it transpires that was a mistake, because not only did organic chemistry win again, but two of the three recipients were carbohydrate chemists by background - my own specialist research area.
The Nobel committee awarded the 2022 prize in equal shares to Barry Sharpless, Morten Meldal and Carolyn Bertozzi, for “an ingenious method of building molecules”, namely the development of click chemistry and bioorthogonal chemistry. The onomatopoeia of ‘click’ chemistry refers to the sound that you make when snapping together blocks of LEGO, and indeed one useful analogy is that click chemistry equates to molecular LEGO; complex molecular pieces can be joined together at will to make larger ones (Fig. 1A).
The basic idea of click chemistry, first proposed by Barry Sharpless more than 20 years ago2, is that using a small selection of ‘perfect’ ‘spring-loaded’ reactions, a modular approach could be used to bolt together molecular building blocks into more complex species. Importantly, each building block contains a mutually reactive handle to ensure that bond formation specifically occurs only as desired. Sharpless then set himself, and other chemists, the goal to develop “a set of powerful, selective, and modular blocks that worked reliably in both small and large-scale applications”. In theory, click chemistry would not only allow the easy construction of molecular complexity by the assembly of modular fragments (Fig. 1B, here the LEGO analogy is even more obvious), but its use would also allow other larger molecular species to be joined together. Highly useful applications involving linking biomacromolecules or surfaces to fluorescent tags (Fig. 1C), or radiolabelling, immediately spring to mind.
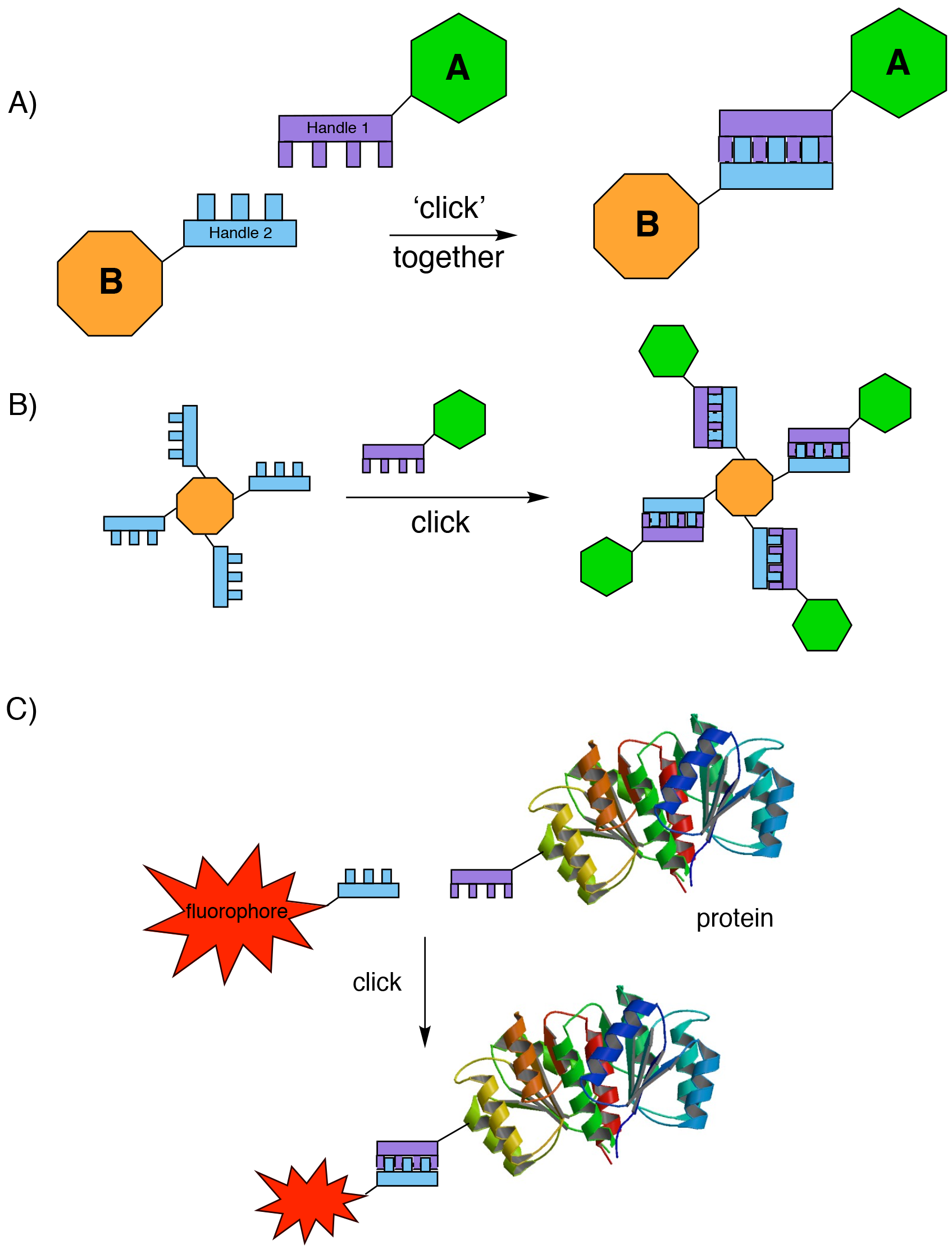
Anyone who has ever heard Sharpless speak knows that his use of the term ‘spring-loaded’ derives from reactions of epoxides; their super reactivity arising from the strain of the three-membered ring, which is released when the epoxide is opened by attack of a nucleophile. Thus the term ‘spring-loaded’ refers to a chemical species that is perfectly primed for reaction, but which will only react with the correct partner. So, in this context ‘perfect’ can be taken to mean that only the desired chemical reaction happens, it irreversibly results in strong covalent bond formation between the two species, and a single predicted product is formed in 100% yield. Finally, the reaction must always work, i.e. it is completely reliable. Oh, and did I add that it also needs to be compatible with aqueous solvents?
In 2000, when Sharpless wrote his concept paper, click chemistry was therefore a nice concept, but unfortunately there was one major problem: in reality there were no ‘perfect’ reactions that satisfied all of the required criteria. You may argue that can’t be true, saying “there are lots of essentially quantitative yielding chemical processes, take amide bond formation for example”. It is indeed true that chemists can make amide bonds in very high yield with excellent reliability. However the problem is that amines also do other reactions, as do the required coupling agents, and you can’t routinely do the coupling reactions in water. So amide bond formation just isn’t good enough to be a click reaction.
However, only just over a year later Medal and Sharpless independently and almost simultaneously found the first example of a perfect click reaction, namely the Cu-catalysed azide-alkyne cycloaddition. It is for the discovery of the catalysed version of this quite old reaction that they were awarded the prize. Very shortly afterwards, Bertozzi would make a key alteration to this reaction that remarkably allowed it to be performed in biological systems; the invention and application of a bioorthogonal click reaction. Let’s now look at the background to this chemical transformation before considering Meldal, Sharpless and then Bertozzi’s contributions in turn.
Background to the Prizes
1,3-Dipolar cycloaddition reactions
Cycloadditions are classic examples of pericyclic reactions. The most well-known is probably the Diels-Alder reaction, familiar to all chemistry undergraduates, which occurs between a diene and an alkene that is formally described as a [4+2] cycloaddition. Perhaps slightly less familiar, though certainly on any undergraduate chemistry curriculum, are 1,3-dipolar [3+2] cycloadditions (Fig, 2A). These encompass a wide range of reactions that occur between a ‘1,3-dipole’ as the first component and an unsaturated species, termed a ‘dipolarophile’, which react together to create a 5-membered ring. A wide range of 1,3-dipoles are known (Fig. 2B), all sharing the common feature that they comprise a conjugated system of three p orbitals containing 4 electrons. While these are neutral species overall, they all contain a separation of charge that is spread over three atoms, hence the term ‘1,3-dipole’. These 1,3-dipoles all undergo cycloaddition reactions with alkenes, alkynes, and other species that contain carbon-heteroatom multiple bonds to produce different types of 5-membered heterocyclic rings systems that are particularly useful in medicinal chemistry.
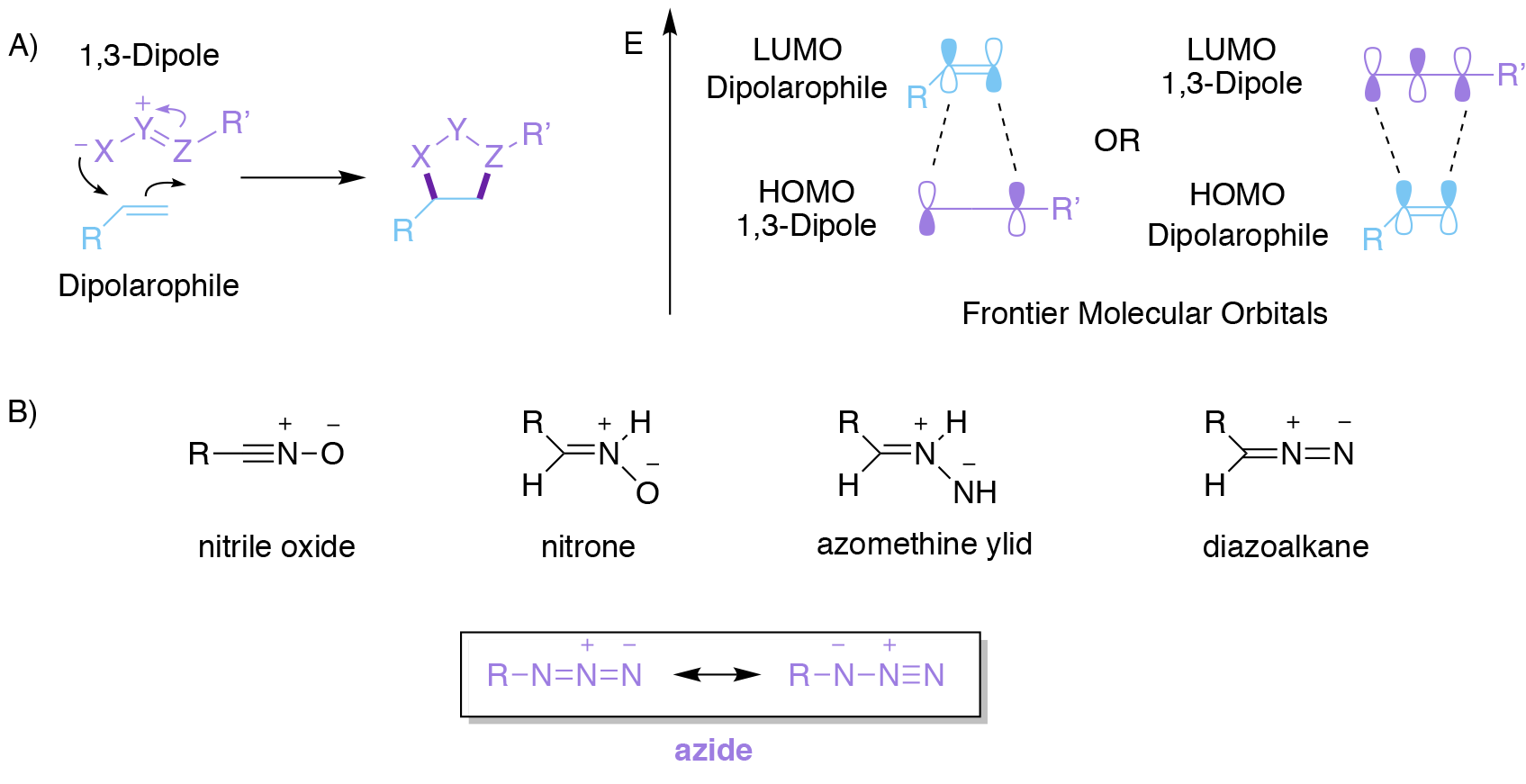
A convenient approach3 to rationalising the rates and regioselectivities of these processes can be made by considering interaction between the Frontier Molecular Orbitals (FMOs) of the two reacting components (Fig. 2A). Broadly speaking, the lower the energy gap between the highest occupied molecular orbital (HOMO) of one component and the lowest unoccupied molecular orbital (LUMO) of the other component, then the better the overlap between these two and the faster the reaction. Whether the most important FMO interaction is between the HOMO of the 1,3-dipole and the LUMO of the dipolarophile (most common, termed normal electron demand), or alternatively between the HOMO of the dipolarophile and the LUMO of the 1,3-dipole (termed inverse electron demand), depends on the precise reaction in question.
Regardless, catalysis may be achieved by decreasing the appropriate HOMO-LUMO energy gap. Furthermore, the better matched that the atom coefficients of the FMOs are, then the more regioselective the reaction tends to be. The 2022 Nobel Prize in Chemistry was awarded for the development of superlative versions of just one of these reactions, namely the azide-alkyne cycloaddition; a reaction that bears the name of its greatest exponent as you can see below.
Rolf Huisgen (1920-2020)4 - ‘creator’ of the 1,3-dioploar cycloaddition

The first account of the cycloaddition reaction of an azide and an alkyne was actually made by Arthur Michael in 1893,5 when he reported the first production of 1,2,3-triazoles. However, the reaction was not studied in any significant detail until Rolf Huisgen, a German organic chemist educated at the University of Munich, began his seminal studies, first at Tubingen University and then at the University of Munich.
Rolf Huisgen’s research interests centred on 1,3-dipolar cycloaddition reactions (Fig. 2). Indeed, it was Huisgen who actually came up the term ‘1,3-dipolar cycloaddition’ in 1960.6 By his meticulous research, Huisgen transformed the entire field, providing unifying rationalisation that seems obvious now, but which no one else had previously appreciated. To quote Albert Szent-Györgyi,7 he “saw what everybody else had seen, but thought what nobody else had thought”. He even predicted new 1,3-dipoles to create new reactions, and went on to carefully study reaction mechanisms, showing that sometimes stepwise rather than concerted processes occurred. A seminal review published by Huisgen in Angewandte Chemie in 1963 summarises these early studies.8 Whilst any of the 1,3-dipolar cycloadditions may be sometimes be referred to as a ‘Huisgen cycloaddition’, it is the azide-alkyne cycloaddition that most commonly bears his name, and our discussion will be limited to this reaction.
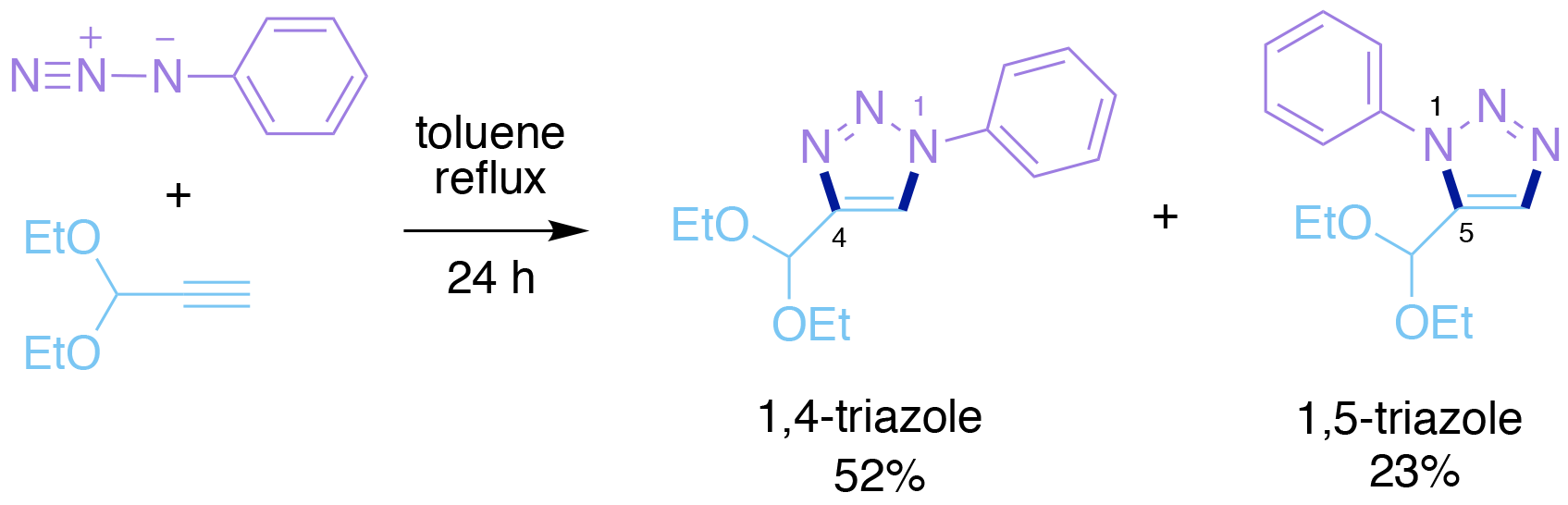
This cycloaddition occurs when an alkyl azide and an alkyne are heated together, to form a triazole product containing a 5-membered heterocyclic ring (Scheme 1). Typical reaction conditions involve refluxing the two components in toluene, i.e. heating to approximately ~110 oC, with the reaction taking 24 h or longer to reach completion. The precise details of this cycloaddition reaction vary with the identities and electronic properties of the two reacting components. However, the reaction is not usually regioselective, i.e. a mixture of two isomeric products is normally formed, namely the 1,4-triazole and 1,5-triazole (Scheme 1). At first such a process therefore appears to be incompletely incompatible with the click chemistry paradigm created by Sharpless. However closer consideration of this reaction reveals the following important details:
a) Neither azides nor alkynes occur significantly9 in biological systems.
b) Both of these functional groups are ‘stable’, and only react with other species under quite specific reaction conditions, none of which occur in biological systems.
c) The Huisgen cycloaddition reaction is completely compatible with aqueous reaction conditions, and its success is generally insensitive to the local environment.
In respect of point b) above it is worth quoting Sharpless himself: “given their feared explosive nature, azides are remarkably stable to water, oxygen, and most conditions used in organic synthesis, until they are exposed to a dipolarophile.”
Thus, while the Huisgen reaction was not a solution, it represented a useful starting point, particularly if methods to enhance the rate of reaction and - most importantly - to completely control product regiochemistry could be found. This sets the scene nicely for the development of effective catalysis of this reaction, the impact of which would be so spectacular that it would merit the award of a Nobel Prize.
The 2022 prize winners
With regard to precise timing of ‘who did it first’, it should be noted that Meldal’s journal paper received date of December 14th 2001 pre-dates that of Sharpless’ work (April 29th 2002) by some 4 months. I shall therefore discuss Meldal’s work first.

Morten Meldal - University of Copenhagen
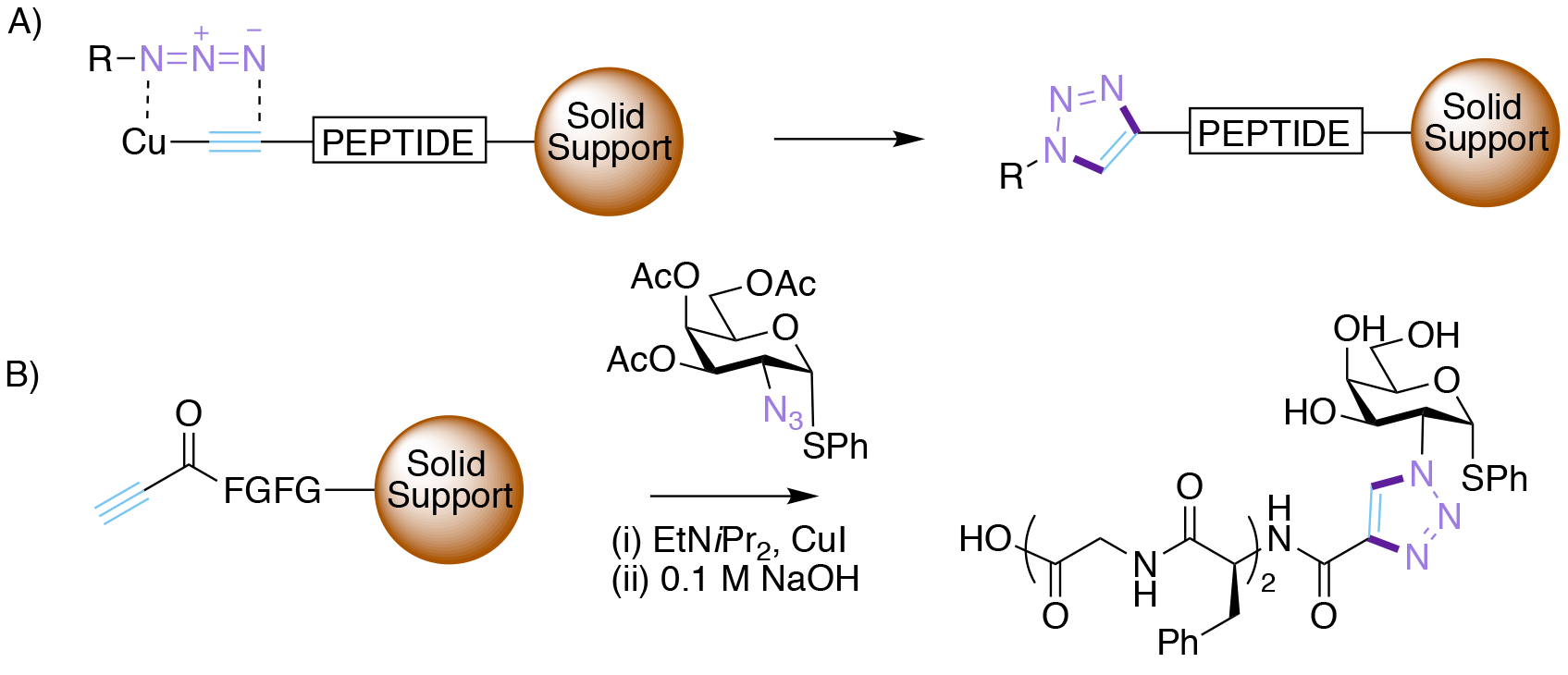
Morten Meldal has a long-standing interest in both carbohydrates, having done his PhD with Klaus Bock at Lyngby, and solid-supported synthesis, the latter particularly when it was applied to combinatorial chemistry. In the key Nobel prize-winning 2002 publication,10 Meldal and his group were seeking to increase the efficiency of the azide-alkyne cycloaddition so that it was compatible with solid-phase combinatorial synthesis of drug molecules. Their goal was to make millions of biologically interesting triazoles available for screening using the split and mix method invented by Furka.11
Meldal’s key reaction insights, based on the earlier literature, were that some metal salts (Na, Mg, Li) of alkynes underwent the azide-alkyne cycloaddition at much lower temperatures, and that one paper, in which the authors were trying to synthesise a propargyl azide using copper (I) chloride, reported unexpected triazole formation. Thus, Medal hypothesised that copper acetylides may be formed by reaction of the alkyne with the Cu (I) salt in the presence of a base (e.g. Hünigs base, N,N-diisoproplyethylamine), and that these copper acetylides would undergo cycloaddition much more readily.
When this hypothesis was explored experimentally the results were spectacular (Scheme 2). Simply adding catalytic amounts of copper (I) salts (chloride, bromide or iodide) to the reaction mixture led to the formation of triazoles in very high yield (>95% conversion), and crucially with complete regiochemical control at 25oC. Interestingly, no ligands were required for the copper. However, Cu (II) salts were ineffective, and the reaction only worked for terminal alkynes. One the other hand the reaction worked exceptionally well on the solid-phase, and was subsequently applied to prepare a wide variety of peptide substrates.
Though the precise mechanism was not investigated at the time, given the accepted precedent that Cu insertion into terminal alkynes occurs in the presence of base (e.g. in the Sonogashira coupling), it was presumed that a copper acetylide intermediate was formed which then underwent either a stepwise or concerted cycloaddition with the azide to form only the 1,4-triazole product.
Barry Sharpless - Scripps Research Institute La Jolla, California

The name Barry Sharpless is probably known to all chemists. He now joins Fred Sanger as only the second person ever to win the Nobel Prize in Chemistry twice, having won a 50% share in 2001 for his development of asymmetric synthesis - the ability to produce single enantiomers of chiral compounds starting from non-chiral starting materials. More specifically he created both the Sharpless asymmetric epoxidation of allylic alcohols and the Sharpless asymmetric dihydroxylation of alkenes. Both of these transformations have long been established parts of the synthetic organic chemist’s repertoire. They are extremely reliable, the outcome is predictable, and which enantiomer of the product is formed can be chosen at will since the chiral catalysts used are readily available.
In the organic chemistry world, Sharpless’ creativity and imagination are therefore legendary. Indeed, some stories around this topic are probably apocryphal, and certainly libellous. What is certain is that in a stroke of creative genius, Sharpless created the concept and coined the phrase ‘click chemistry.’ Next, he went on to translate his concept into a working reality by the independent discovery of Cu (I) catalysis of the Huisgen azide-alkyne cycloaddition.
Sharpless’s group’s12-13 - seminal publication on Cu (I) catalysis of the azide-alkyne cycloaddition appeared in print shortly after Meldal’s. In this paper, which has now been cited more than 13,000 times, Sharpless reported that the Cu (I) catalyst was best made by in situ reduction of Cu (II) salts, such as CuSO4 hydrate, by ascorbic acid, or sodium ascorbate. As little as 1 mol % of the catalyst was sufficient. The reaction was ‘extremely forgiving’, did not require any special conditions or precautions, was complete at room temperature after 6-36 h, and worked in a range of solvents, most importantly water without any added organic co-solvent. Thus, the reaction had wide scope and functional group tolerance, and was essentially ‘fool-proof.’
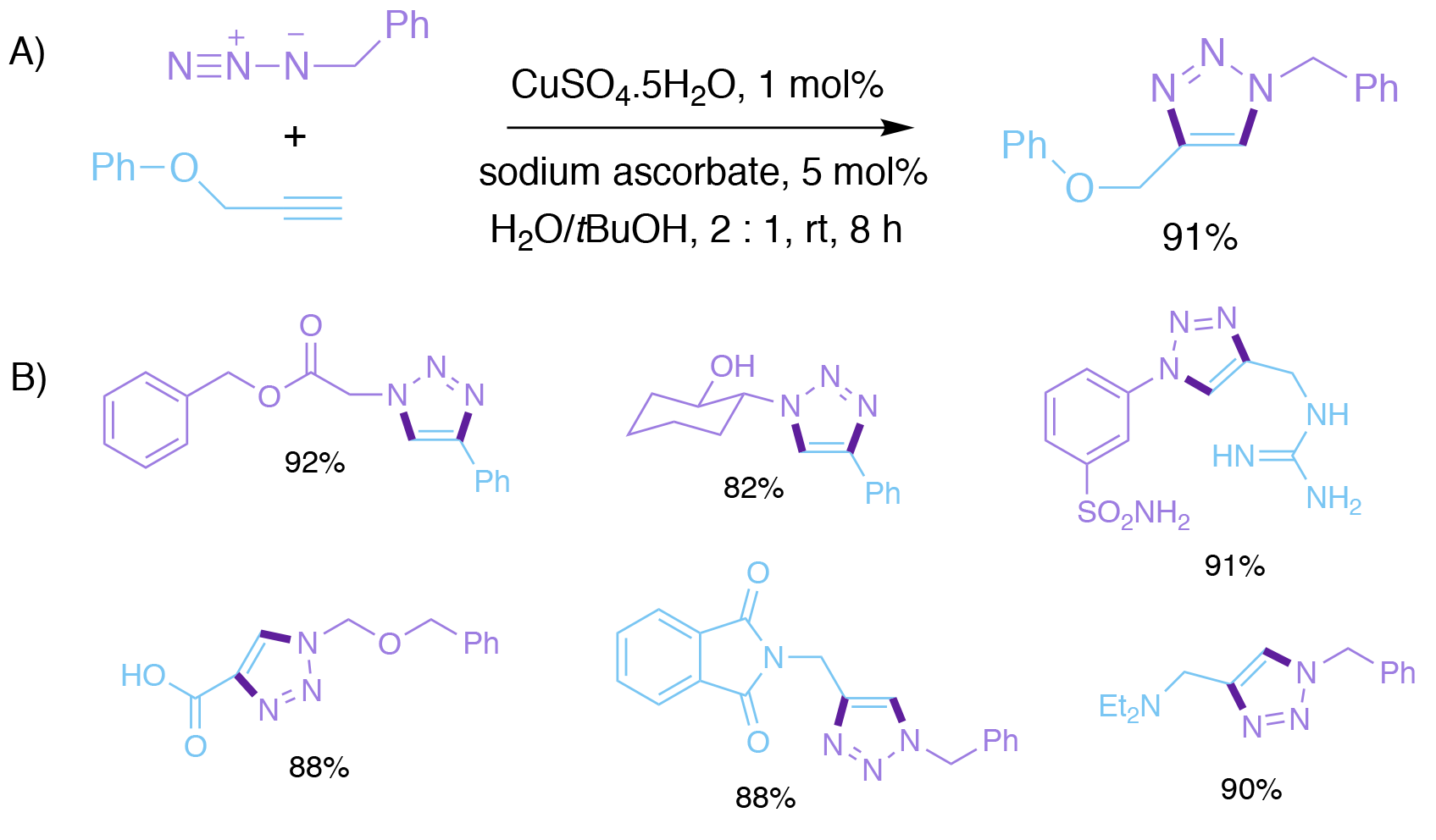
Scheme 3. Sharpless and Fokin’s Cu-catalysed azide-alkyne cycloaddition. A) Generation of the Cu (I) catalyst in situ by reduction of CuSO4 with sodium ascorbate. B) Representative examples.
In this paper, Sharpless and Fokin went on to propose a catalytic cycle for the Cu(I)-catalysed ligation reaction, which involved an intermediate Cu acetylide. Based on density functional theory (DFT) calculations, a step-wise mechanism was proposed instead of a concerted cycloaddition process. Later studies by Fokin and Finn would reveal that two Cu(I) ions were involved, and that the mechanism was slightly more complicated than originally proposed.14
Despite these minor uncertainties, one thing was sure; the first ‘perfect’ click reaction had been discovered. Although Meldal’s reaction conditions using a Cu (I) additive were reported first, it is the use of Cu (II) and its in situ reduction to the active Cu (I) species by sodium ascorbate, that has become the almost exclusively applied procedure. Click chemistry had become a reality, using a simple reaction that could be performed by anyone, anywhere and at any time.
Carolyn Bertozzi - Stanford University, California

Like Meldal, Carolyn Bertozzi is a carbohydrate chemist by background, having done her PhD with Mark Bednarski at Berkley on the chemical synthesis of C glycosides. Realising that sugars are of vital importance throughout biological systems, she embarked on her own research programs at the interface of chemistry and biology. The genius of Bertozzi was to recognise the golden opportunities of applying synthetic chemistry techniques directly in biological systems, and click chemistry ultimately provided an excellent means of functionalising biomolecules in living systems.
Bertozzi developed the concept and coined the term ‘biorthogonal chemistry’ in 2003,15 which is defined as a chemical reaction that can occur within living cells, which neither affects, nor is affected by, the myriad of native biochemical species and metabolic processes that may also be present. It doesn’t take much of an intuitive leap to then see that click chemistry, as defined by Sharpless, provides the perfect basis for bioorthogonal chemistry. Indeed the case that Bertozzi made for the using biorthogonal click chemistry to study biological systems was extremely compelling. For example, would it not be extremely useful if one reacting handle of a click chemistry pair could be incorporated into a cell, and then the other reacting handle used as a probe to find its location? Furthermore, attaching a label such as a fluorophore to a ‘clickable’ probe would allow techniques such as fluorescence microscopy to be used to visualise the whole process in living cells.
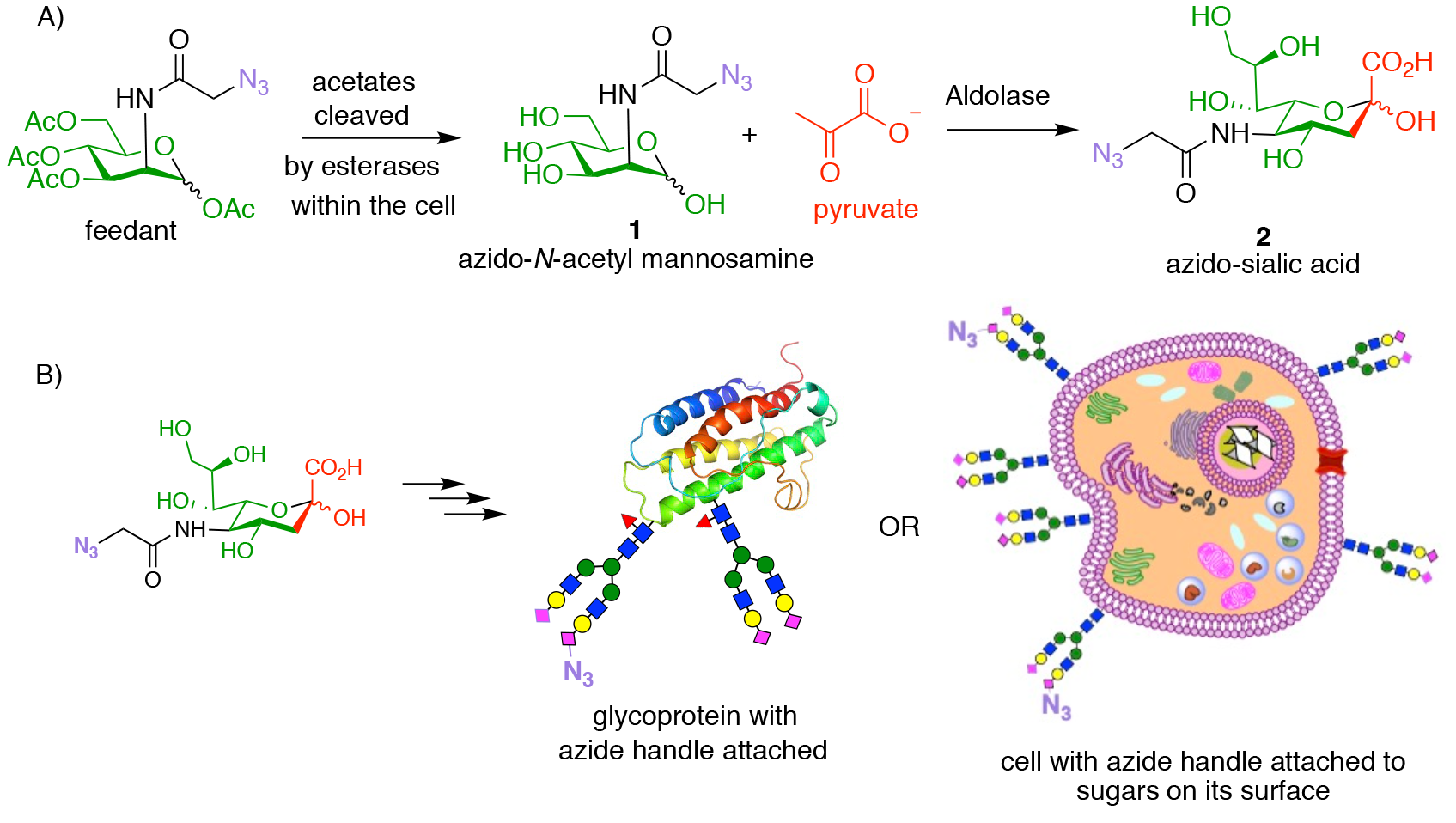
Azide, which is not itself found within biological systems and displays essentially no reactivity towards biological systems or metabolites, appeared to be the perfect reactive handle with which to perform bioorthogonal chemistry. The first key development by Bertozzi was the demonstration that reactive azide handles can be incorporated either into recombinant proteins, and even onto the surface of live cells, simply by feeding cells with a peracetylated azido N-acetylmannosamine sugar (Fig. 3A, feedant). This is possible because the sugar sialic acid is found at the extremities of the sugar chains (glycans, or oligosaccharides) attached to mammalian proteins and to mammalian cell surfaces. Sialic acid is biosynthesised within cells from N-acetylmannosamine and pyruvate (shown in red) in an aldol reaction. However, the enzymes that catalyse this process are promiscuous and also accept azido N-acetyl mannosamine 1 (formed inside the cell following loss of the acetate groups from the peracetylated feedstock) as a substrate, to produce azido-sialic acid 2. Thus, when the surrogate azide containing substrate is fed to cells, some of the sialic acids that it makes will then contain an azide. These sialic acids are further metabolically processed to occupy some of the positions throughout the cell that sialic acid normally occupies, and so provide bioorthogonal reactive handles to potentially carry out click chemistry.
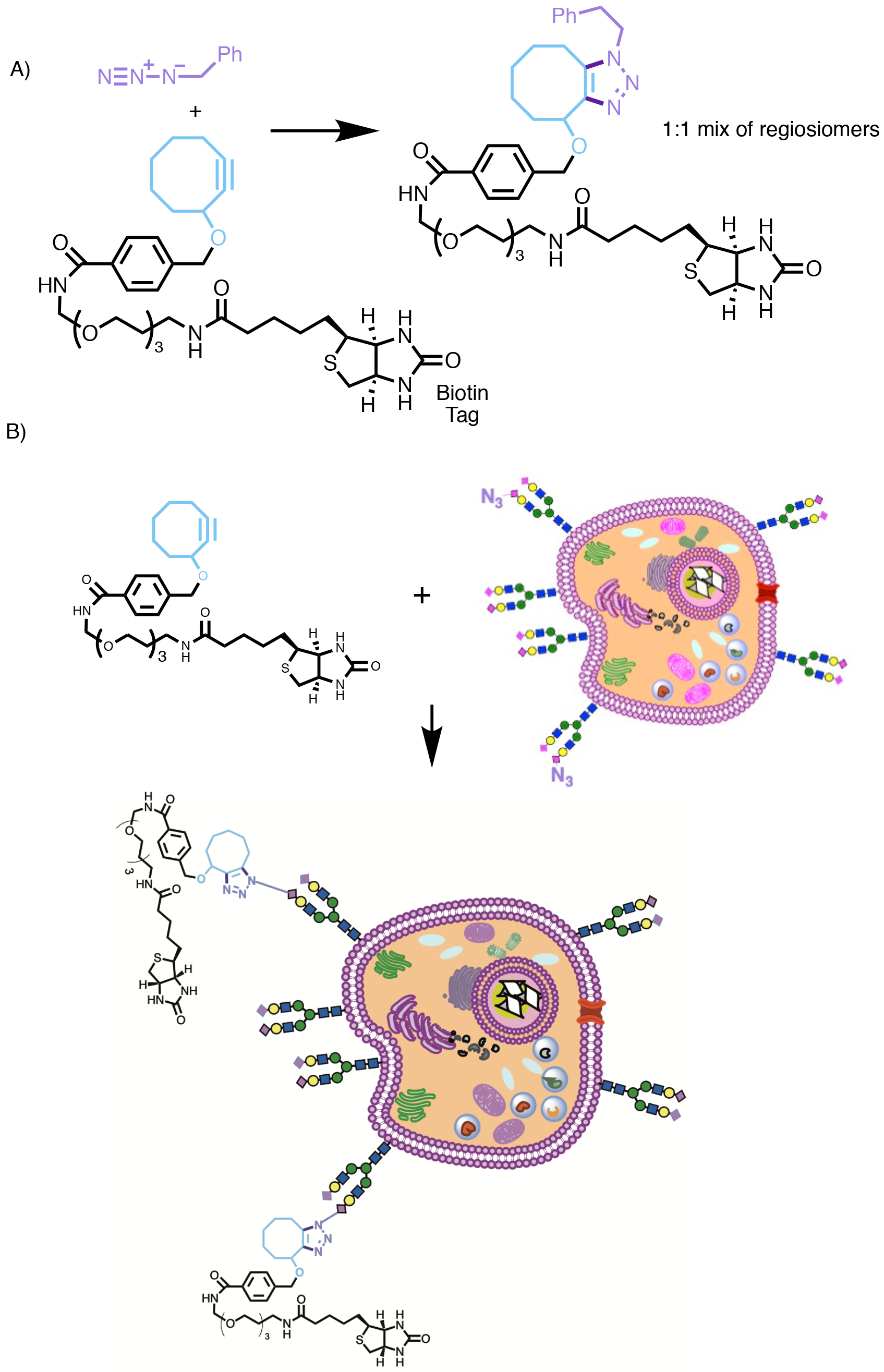
Given this ability to incorporate azide into living systems, the opportunity for selective reaction of azide in order to attach externally administered chemical species became possible. Although Bertozzi had developed other approaches to bioorthogonally react azide, such as the Staudinger ligation,16 the Cu-catalysed variant of the Huisgen cycloaddition developed by Meldal and Sharpless seemed like the perfect reaction to apply.17 However, there was one big problem; the copper. Copper is toxic, and so cannot be used in cells or living organisms. Furthermore, often the reaction still required heating to a moderate temperature in order to increase its rate, with reactions often taking as long as 36 h at room temperature. So, in order to develop a version of the Huisgen cycloaddition that could be applied directly in biological systems something else was needed. Bertozzi therefore very wisely did something that far too few of us academics ever do; she looked back into the old chemical literature.18 She then realised that putting an alkyne in a medium sized ring greatly increased its reactivity and so here was a potential solution to the Cu toxicity and rate of reaction problems.
In the first example reported (Scheme 4), Bertozzi used a cyclooctyne as the dipolarophile. An 8-membered ring is the smallest ring into which an alkyne may be incorporated to form a stable product.19 In this instance the cyclooctyne was attached via a short linker to a biotin ‘handle’, which was attached as a reporting unit to enable confirmation that subsequent reaction had occurred later on using anti-biotin antibody and Western Blotting techniques. In this way, Bertozzi was able to demonstrate that click reactions had occurred successfully, first using small molecule models (Scheme 4A, note the reaction is not regioselective but this is of no consequence), then a recombinant glycoprotein produced in Chinese Hamster Ovary (CHO) cells, and finally demonstrating that click reactions had occurred on the surface of Jurkat cells (Scheme 4B, immortalised human T lymphocyte cells). Later refinements were made to the alkyne component,20 including the introduction of two election withdrawing fluorines on the cyclooctyne ring (to increase the rate of cycloaddition with azide by lowering the energy of the LUMO of the alkyne), and the development of a water soluble hydrophilic cyclooctyne suitable for reactions in aqueous biological systems.21
Some thoughts on the prize awards
Morten Meldal’s group’s seminal publication was quite remarkably published in the Journal of Organic Chemistry in 2002, commonly referred to by organic chemists as ‘JOC’. Without meaning any disrespect to the editors of, or authors who publish excellent work in JOC, this is in a ‘lower tier’ of American Chemical Society (ACS) journals and currently has an impact factor of only 4.3. One wonders if this was indeed the journal that Meldal submitted his work to, or perhaps if a journal editor of a higher-ranked ACS journal declined to publish the paper saying it was ‘too specialised’ or ‘not of interest to our broad readership’? This is of course mere speculation on my part, and perhaps JOC was the first and only destination for this particular publication.
However, like many other chemists who fight against perceptions of ‘relative importance’ during both editorial ‘sifting’ and then the anonymous peer review process, I am heartened that the Nobel Committee recognised this work for its real value, and that one can be awarded a Nobel prize essentially for a single publication in a middle tier journal such as JOC.
Furthermore, anyone who has studied the history of science in detail, or, perhaps rather like me who has read Bill Bryson’s excellent book, A Short History of Nearly Everything23 will be all too aware that throughout history often the real inventors or discoveries of processes have often been forgotten in favour of more ‘popular’ or ‘more powerful’ figures who have grabbed all of the limelight. It is particularly pleasing that Morten Meldal was awarded his fair share of this years’ prize, despite the fact that the Nobel laureate Sharpless essentially made the same discovery at the same time.
The rise of click chemistry
Database searches and citation counts give some idea of the impact of a field. A Scopus search (October 2022) for the term ‘click’ in article titles, keywords or abstracts produced >49,000 hits, with approximately 3,500 hits per year for the years 2019, 2020 and 2021. This massive citation count is unsurprising, since the beauty of click chemistry, as Sharpless originally envisioned, is that it can basically be applied anywhere, anytime, and by anyone. The reaction is by design so robust and insensitive to other reagents or functional groups, that it doesn’t matter what the substrates are; the reaction just works. Furthermore, the reaction works well in water and is insensitive to oxygen, so basically anyone can do it (even biologists 😊); prior expertise in synthetic organic chemistry is absolutely not required!
Therefore, it is no surprise that the Cu(I)-catalysed azide-alkyne cycloaddition has now been applied to almost every situation that one can imagine where the objective has been to join two molecules linked together, including (but not limited to) pharmaceutical development and drug discovery, conjugation of tags to biomolecules, DNA sequencing and throughout materials science. It is unfortunately not possible in an article such as this to begin to scratch the surface of the myriad applications of this truly remarkably robust reaction, though many excellent reviews are available.24 Even my own research group jumped on the ‘click bandwagon’, so to speak, and developed an application of click chemistry to allow the direct conjugation of unprotected sugars to other species in aqueous solvent systems.25 Other reactions have been promulgated as ‘click’ chemistry, for example the thiol-ene reaction which comprises the formal addition of a thiol to an alkene, and proceeds either by free radical or Michael-type mechanisms. However, none of these other reactions matches the performance of the Cu(I)-catalysed azide-alkyne cycloaddition. Therefore, to the majority of chemists the Cu(I)-catalysed Huisgen cycloaddition remains the pre-eminent example of click chemistry, and indeed it is widely regarded as synonymous with the term ‘click reaction’.
The rise of bioorthogonal chemistry
Whilst the concept of biorthogonality is by definition less widely applicable than click chemistry (a Scopus search produces a mere 2,500 hits), it provides chemists with unique methods to study biomolecule interactions inside cells, and crucially also to decipher disease and disease processes. Indeed, bioorthogonal chemistry has become one of the cornerstones of chemical biology research. For the uninitiated, chemical biology is the application of chemistry, and in particular synthesis, to study and manipulate biological systems. Clearly a chemical reaction that you can perform in water, in the presence of oxygen, that links together two molecules containing mutually reactive handles in essentially quantitative yield is rather useful for the study of biology! Moreover, if you can even carry out click chemsitry in living cells, while looking at the cell through a microscope at the same time, then so much the better. Unfortunately, again there is not space in this article to even begin to do justice to the field of bioorthogonal chemistry, and the interested reader is therefore strongly encouraged to consult one of the many excellent specialist reviews on this subject.26
Afterthoughts
Sharpless is a creative genius, who absolutely deserves his pre-eminence in the chemistry world by the award of a second Nobel Prize. It was he who created the click chemistry concept, but it was Meldal who discovered the first example, pipping Sharpless at the post by a few months. Bertozzi is another creative genius, who furthermore possessed the insight, courage and drive to tackle the toughest, but perhaps the most rewarding, of problems; how to do synthetic chemistry in living biological systems. All three are truly worthy winners of the 2022 Nobel Prize in Chemistry and I congratulate them wholeheartedly.
Rolf Huisgen’s death in 2020, at the amazing age of 99, was ultimately untimely, not least because he fell short of a remarkable century by only three months. Astonishingly Huisgen had continued to publish until 2018.27 He was a true giant of organic chemistry, and it is fitting that one of his numerous legacies was the fundamental reaction for which the 2022 Nobel Prize was awarded. Had he still been alive the Nobel Committee may have been faced with a real conundrum as to who to omit from this year’s prize, as there can only be three winners. One also wonders whether without the seminal work of Huisgen, would either Sharpless or Meldal would have focussed on that particular cycloaddition reaction as a means of linking two molecules together? On the other hand, Bertozzi had already developed her first bioorthogonal reaction - the Staudinger ligation - so the future development of biorthogonal chemistry was in some respects already assured.
Without Huisgen one also wonders if alternative solutions would have been found, or in fact whether they even exist? It is notable that, 20 years after the first reports, the Cu-catalysed and strain promoted azide-alkyne cycloaddition reactions are still pre-eminent over all other click reactions that have been developed; nothing else actually comes close. The final question therefore is, “Are there other click reactions out there that are yet to be discovered?” We shall have to wait and see.
Finally I conclude in a similar vein to last year.28 Katalin Karikó and Drew Weissman surely must win the Nobel Prize in Chemistry for their work on mRNA vaccines at some not too distant point in the future. Maybe 2023 will be their year? If so, then I may be back again to complete a trilogy of articles.
References and notes
- https://www.youtube.com/watch?v=-Ch3VJhIbH4
- Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem. Int. Ed. 2001, 40, 2004–2021.
- Fleming, I. 2010, Molecular Orbitals and Organic Chemical Reactions, Reference Edition, John Wiley and Sons.
- For an Obituary see: Trauner, D. Nat. Chem. Biol. 2020, 16, 711.
- Michael, A. J. Prakt. Chem. 1893, 48, 94.
- R. Huisgen, Proc. Chem. Soc. London 1961, 357–369.
- A Hungarian Biochemist who won the Nobel Prize for Medicine and Physiology in 1937.
- Huisgen, R. Angew. Chem. Int. Ed. 1963, 2, 565-632.
- Whilst of limited occurrence, alkynes are present in some natural products, and are formed during some biosynthetic pathways. For a recent review see: X. Li, J.-M. Lv, D. Hu, I. Abe, RSC Chem. Biol. 2021, 2, 166–180. However, to the best of my knowledge, azides are not found anywhere in natural biological systems.
- Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064.
- Furka Á.; Sebestyén. F.; Asgedom, M.; Dibó, G. Int. J. Peptide Protein Res. 1991, 37, 487-493.
- Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem. Int. Ed. 2002, 41, 2596–2599.
- It is noted that Valery Fokin, a Postdoc in Sharpless’ group at the time, is joint corresponding author of this paper.
- Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem. Int. Ed. 2005, 44, 2210–2215.
- Hang, H. C.; Yu, C.; Kato, D. L.; Bertozzi, C. R. Proc. Natl. Acad. Sci. USA 2003, 100, 14846–14851.
- Saxon, E.; Bertozzi, C. R. Science 2000, 287, 2007–2010.
- Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272–1279.
- Wittig, G.; Krebs, A. Chem. Ber. 1961, 94, 3260–3275.
- Agard, N. J.; Prescher, J. A.; Bertozzi, C. R. J. Am. Chem. Soc. 2004, 126, 15046–15047.
- Agard, N. J.; Baskin, J. M.; Prescher, J. A.; Lo, A.; Bertozzi, C. R. ACS Chem. Biol. 2006, 1 (10), 644–648.
- Sletten, E. M.; Bertozzi, C. R. Org. Lett. 2008, 10, 3097–3099.
- Baskin, J. M.; Prescher, J. A.; Laughlin, S. T.; Agard, N. J.; Chang, P. V.; Miller, I. A.; Lo, A.; Codelli, J. A.; Bertozzi, C. R. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797.
- Bryson, B. 2003, A short history of nearly everything, New York, Broadway Books.
- a) Kaur, J.; Saxena, M.; Rishi, N. Bioconjug. Chem. 2021, 32, 1455–1471; b) Castro, V.; Rodríguez, H.; Albericio, F. ACS Comb. Sci. 2016, 18, 1–14; c) Tiwari, V. K.; Mishra, B. B.; Mishra, K. B.; Mishra, N.; Singh, A. S.; Chen, X. Chem. Rev. 2016, 116, 3086–3240; d) Poonthiyil, V.; Lindhorst, T. K.; Golovko, V. B.; Fairbanks, A. J. Beilstein J. Org. Chem. 2018, 14, 11–24.
- Lim, D.; Brimble, M. A.; Kowalczyk, R.; Watson, A. J. A.; Fairbanks, A. J. Angew. Chem. Int. Ed. 2014, 53, 11907–11911.
- For some examples see: a) Scinto, S. L.; Bilodeau, D. A.; Hincapie, R.; Lee, W.; Nguyen, S. S.; Xu, M.; am Ende, C. W.; Finn, M. G.; Lang, K.; Lin, Q.; Pezacki, J. P.; Prescher, J. A.; Robillard, M. S.; Fox, J. M. Nat. Rev. Methods Primers 2021, 1, 1-23; b) Taiariol, L.; Chaix, C.; Farre, C.; Moreau, E. Chem. Rev. 2022, 122, 340–384; c) Wu, D.; Yang, K.; Zhang, Z.; Feng, Y.; Rao, L.; Chen, X.; Yu, G. Chem. Soc. Rev. 2022, 51, 1336–1373; d) Stump, B. ChemBioChem 2022, 23, e202200016.
- Breugst, M.; Huisgen, R.; Reissig, H. U. Eur. J. Org. Chem. 2018, 2477–2485.
- Fairbanks, A. J. Chem. NZ 2022, 86, 10-14
.jpg)


